 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Spatio-Temporal SE(3) Diffusion for Long-Horizon Protein Dynamics
Feb 02, 2026Molecular dynamics (MD) simulations remain the gold standard for studying protein dynamics, but their computational cost limits access to biologically relevant timescales. Recent generative models have shown promise in accelerating simulations, yet they struggle with long-horizon generation due to architectural constraints, error accumulation, and inadequate modeling of spatio-temporal dynamics. We present STAR-MD (Spatio-Temporal Autoregressive Rollout for Molecular Dynamics), a scalable SE(3)-equivariant diffusion model that generates physically plausible protein trajectories over microsecond timescales. Our key innovation is a causal diffusion transformer with joint spatio-temporal attention that efficiently captures complex space-time dependencies while avoiding the memory bottlenecks of existing methods. On the standard ATLAS benchmark, STAR-MD achieves state-of-the-art performance across all metrics--substantially improving conformational coverage, structural validity, and dynamic fidelity compared to previous methods. STAR-MD successfully extrapolates to generate stable microsecond-scale trajectories where baseline methods fail catastrophically, maintaining high structural quality throughout the extended rollout. Our comprehensive evaluation reveals severe limitations in current models for long-horizon generation, while demonstrating that STAR-MD's joint spatio-temporal modeling enables robust dynamics simulation at biologically relevant timescales, paving the way for accelerated exploration of protein function.
Simultaneous Modeling of Protein Conformation and Dynamics via Autoregression
May 23, 2025Understanding protein dynamics is critical for elucidating their biological functions. The increasing availability of molecular dynamics (MD) data enables the training of deep generative models to efficiently explore the conformational space of proteins. However, existing approaches either fail to explicitly capture the temporal dependencies between conformations or do not support direct generation of time-independent samples. To address these limitations, we introduce ConfRover, an autoregressive model that simultaneously learns protein conformation and dynamics from MD trajectories, supporting both time-dependent and time-independent sampling. At the core of our model is a modular architecture comprising: (i) an encoding layer, adapted from protein folding models, that embeds protein-specific information and conformation at each time frame into a latent space; (ii) a temporal module, a sequence model that captures conformational dynamics across frames; and (iii) an SE(3) diffusion model as the structure decoder, generating conformations in continuous space. Experiments on ATLAS, a large-scale protein MD dataset of diverse structures, demonstrate the effectiveness of our model in learning conformational dynamics and supporting a wide range of downstream tasks. ConfRover is the first model to sample both protein conformations and trajectories within a single framework, offering a novel and flexible approach for learning from protein MD data.
ProteinBench: A Holistic Evaluation of Protein Foundation Models
Sep 10, 2024Recent years have witnessed a surge in the development of protein foundation models, significantly improving performance in protein prediction and generative tasks ranging from 3D structure prediction and protein design to conformational dynamics. However, the capabilities and limitations associated with these models remain poorly understood due to the absence of a unified evaluation framework. To fill this gap, we introduce ProteinBench, a holistic evaluation framework designed to enhance the transparency of protein foundation models. Our approach consists of three key components: (i) A taxonomic classification of tasks that broadly encompass the main challenges in the protein domain, based on the relationships between different protein modalities; (ii) A multi-metric evaluation approach that assesses performance across four key dimensions: quality, novelty, diversity, and robustness; and (iii) In-depth analyses from various user objectives, providing a holistic view of model performance. Our comprehensive evaluation of protein foundation models reveals several key findings that shed light on their current capabilities and limitations. To promote transparency and facilitate further research, we release the evaluation dataset, code, and a public leaderboard publicly for further analysis and a general modular toolkit. We intend for ProteinBench to be a living benchmark for establishing a standardized, in-depth evaluation framework for protein foundation models, driving their development and application while fostering collaboration within the field.
Protein Conformation Generation via Force-Guided SE(3) Diffusion Models
Mar 21, 2024
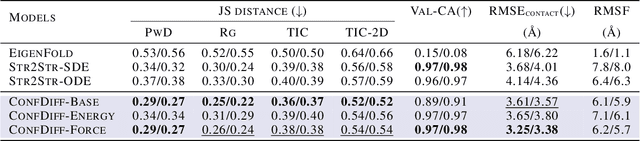
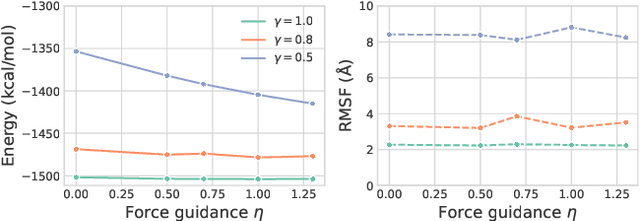
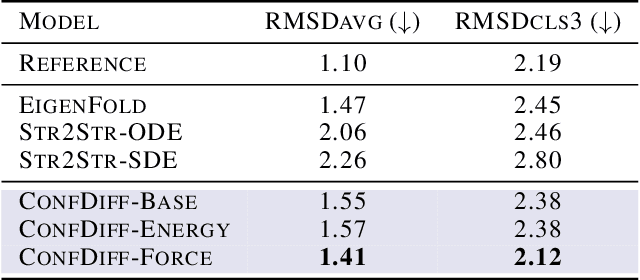
The conformational landscape of proteins is crucial to understanding their functionality in complex biological processes. Traditional physics-based computational methods, such as molecular dynamics (MD) simulations, suffer from rare event sampling and long equilibration time problems, hindering their applications in general protein systems. Recently, deep generative modeling techniques, especially diffusion models, have been employed to generate novel protein conformations. However, existing score-based diffusion methods cannot properly incorporate important physical prior knowledge to guide the generation process, causing large deviations in the sampled protein conformations from the equilibrium distribution. In this paper, to overcome these limitations, we propose a force-guided SE(3) diffusion model, ConfDiff, for protein conformation generation. By incorporating a force-guided network with a mixture of data-based score models, ConfDiff can can generate protein conformations with rich diversity while preserving high fidelity. Experiments on a variety of protein conformation prediction tasks, including 12 fast-folding proteins and the Bovine Pancreatic Trypsin Inhibitor (BPTI), demonstrate that our method surpasses the state-of-the-art method.
Learning Harmonic Molecular Representations on Riemannian Manifold
Mar 27, 2023Molecular representation learning plays a crucial role in AI-assisted drug discovery research. Encoding 3D molecular structures through Euclidean neural networks has become the prevailing method in the geometric deep learning community. However, the equivariance constraints and message passing in Euclidean space may limit the network expressive power. In this work, we propose a Harmonic Molecular Representation learning (HMR) framework, which represents a molecule using the Laplace-Beltrami eigenfunctions of its molecular surface. HMR offers a multi-resolution representation of molecular geometric and chemical features on 2D Riemannian manifold. We also introduce a harmonic message passing method to realize efficient spectral message passing over the surface manifold for better molecular encoding. Our proposed method shows comparable predictive power to current models in small molecule property prediction, and outperforms the state-of-the-art deep learning models for ligand-binding protein pocket classification and the rigid protein docking challenge, demonstrating its versatility in molecular representation learning.