 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Behavioral Alignment in LLM Social Simulations via Context Formation and Navigation
Jan 04, 2026Large language models (LLMs) are increasingly used to simulate human behavior in experimental settings, but they systematically diverge from human decisions in complex decision-making environments, where participants must anticipate others' actions and form beliefs based on observed behavior. We propose a two-stage framework for improving behavioral alignment. The first stage, context formation, explicitly specifies the experimental design to establish an accurate representation of the decision task and its context. The second stage, context navigation, guides the reasoning process within that representation to make decisions. We validate this framework through a focal replication of a sequential purchasing game with quality signaling (Kremer and Debo, 2016), extending to a crowdfunding game with costly signaling (Cason et al., 2025) and a demand-estimation task (Gui and Toubia, 2025) to test generalizability across decision environments. Across four state-of-the-art (SOTA) models (GPT-4o, GPT-5, Claude-4.0-Sonnet-Thinking, DeepSeek-R1), we find that complex decision-making environments require both stages to achieve behavioral alignment with human benchmarks, whereas the simpler demand-estimation task requires only context formation. Our findings clarify when each stage is necessary and provide a systematic approach for designing and diagnosing LLM social simulations as complements to human subjects in behavioral research.
PsyLite Technical Report
Jun 26, 2025With the rapid development of digital technology, AI-driven psychological counseling has gradually become an important research direction in the field of mental health. However, existing models still have deficiencies in dialogue safety, detailed scenario handling, and lightweight deployment. To address these issues, this study proposes PsyLite, a lightweight psychological counseling large language model agent developed based on the base model InternLM2.5-7B-chat. Through a two-stage training strategy (hybrid distillation data fine-tuning and ORPO preference optimization), PsyLite enhances the model's deep-reasoning ability, psychological counseling ability, and safe dialogue ability. After deployment using Ollama and Open WebUI, a custom workflow is created with Pipelines. An innovative conditional RAG is designed to introduce crosstalk humor elements at appropriate times during psychological counseling to enhance user experience and decline dangerous requests to strengthen dialogue safety. Evaluations show that PsyLite outperforms the baseline models in the Chinese general evaluation (CEval), psychological counseling professional evaluation (CPsyCounE), and dialogue safety evaluation (SafeDialBench), particularly in psychological counseling professionalism (CPsyCounE score improvement of 47.6\%) and dialogue safety (\safe{} score improvement of 2.4\%). Additionally, the model uses quantization technology (GGUF q4\_k\_m) to achieve low hardware deployment (5GB memory is sufficient for operation), providing a feasible solution for psychological counseling applications in resource-constrained environments.
The Value of AI-Generated Metadata for UGC Platforms: Evidence from a Large-scale Field Experiment
Dec 24, 2024



AI-generated content (AIGC), such as advertisement copy, product descriptions, and social media posts, is becoming ubiquitous in business practices. However, the value of AI-generated metadata, such as titles, remains unclear on user-generated content (UGC) platforms. To address this gap, we conducted a large-scale field experiment on a leading short-video platform in Asia to provide about 1 million users access to AI-generated titles for their uploaded videos. Our findings show that the provision of AI-generated titles significantly boosted content consumption, increasing valid watches by 1.6% and watch duration by 0.9%. When producers adopted these titles, these increases jumped to 7.1% and 4.1%, respectively. This viewership-boost effect was largely attributed to the use of this generative AI (GAI) tool increasing the likelihood of videos having a title by 41.4%. The effect was more pronounced for groups more affected by metadata sparsity. Mechanism analysis revealed that AI-generated metadata improved user-video matching accuracy in the platform's recommender system. Interestingly, for a video for which the producer would have posted a title anyway, adopting the AI-generated title decreased its viewership on average, implying that AI-generated titles may be of lower quality than human-generated ones. However, when producers chose to co-create with GAI and significantly revised the AI-generated titles, the videos outperformed their counterparts with either fully AI-generated or human-generated titles, showcasing the benefits of human-AI co-creation. This study highlights the value of AI-generated metadata and human-AI metadata co-creation in enhancing user-content matching and content consumption for UGC platforms.
Efficient and generalizable prediction of molecular alterations in multiple cancer cohorts using H&E whole slide images
Jul 22, 2024Molecular testing of tumor samples for targetable biomarkers is restricted by a lack of standardization, turnaround-time, cost, and tissue availability across cancer types. Additionally, targetable alterations of low prevalence may not be tested in routine workflows. Algorithms that predict DNA alterations from routinely generated hematoxylin and eosin (H&E)-stained images could prioritize samples for confirmatory molecular testing. Costs and the necessity of a large number of samples containing mutations limit approaches that train individual algorithms for each alteration. In this work, models were trained for simultaneous prediction of multiple DNA alterations from H&E images using a multi-task approach. Compared to biomarker-specific models, this approach performed better on average, with pronounced gains for rare mutations. The models reasonably generalized to independent temporal-holdout, externally-stained, and multi-site TCGA test sets. Additionally, whole slide image embeddings derived using multi-task models demonstrated strong performance in downstream tasks that were not a part of training. Overall, this is a promising approach to develop clinically useful algorithms that provide multiple actionable predictions from a single slide.
Enhancing Instance-Level Image Classification with Set-Level Labels
Nov 17, 2023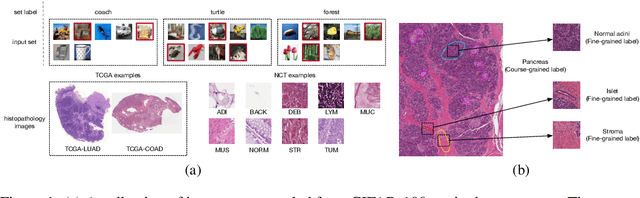

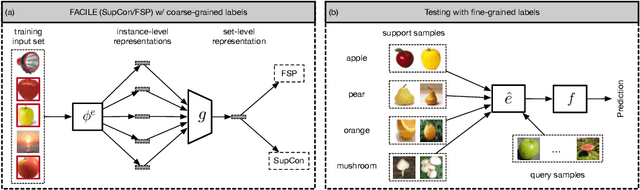
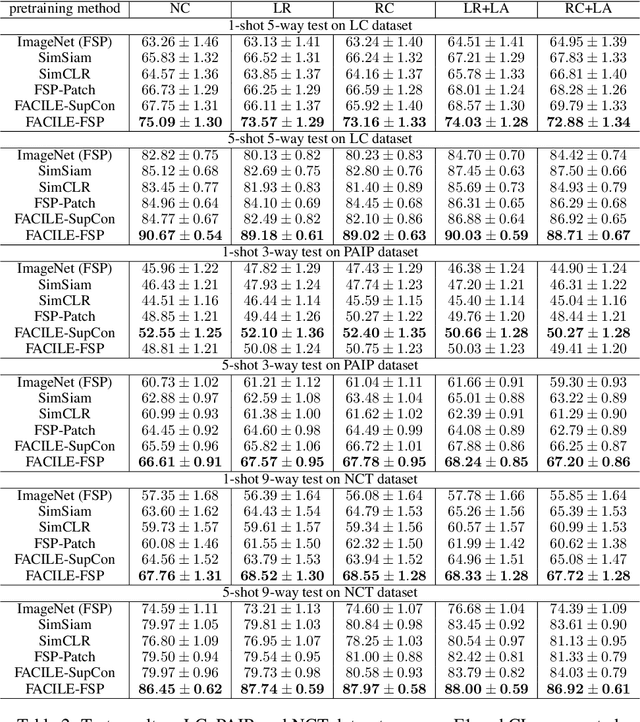
Instance-level image classification tasks have traditionally relied on single-instance labels to train models, e.g., few-shot learning and transfer learning. However, set-level coarse-grained labels that capture relationships among instances can provide richer information in real-world scenarios. In this paper, we present a novel approach to enhance instance-level image classification by leveraging set-level labels. We provide a theoretical analysis of the proposed method, including recognition conditions for fast excess risk rate, shedding light on the theoretical foundations of our approach. We conducted experiments on two distinct categories of datasets: natural image datasets and histopathology image datasets. Our experimental results demonstrate the effectiveness of our approach, showcasing improved classification performance compared to traditional single-instance label-based methods. Notably, our algorithm achieves 13% improvement in classification accuracy compared to the strongest baseline on the histopathology image classification benchmarks. Importantly, our experimental findings align with the theoretical analysis, reinforcing the robustness and reliability of our proposed method. This work bridges the gap between instance-level and set-level image classification, offering a promising avenue for advancing the capabilities of image classification models with set-level coarse-grained labels.
An Adam-enhanced Particle Swarm Optimizer for Latent Factor Analysis
Feb 23, 2023Digging out the latent information from large-scale incomplete matrices is a key issue with challenges. The Latent Factor Analysis (LFA) model has been investigated in depth to an alyze the latent information. Recently, Swarm Intelligence-related LFA models have been proposed and adopted widely to improve the optimization process of LFA with high efficiency, i.e., the Particle Swarm Optimization (PSO)-LFA model. However, the hyper-parameters of the PSO-LFA model have to tune manually, which is inconvenient for widely adoption and limits the learning rate as a fixed value. To address this issue, we propose an Adam-enhanced Hierarchical PSO-LFA model, which refines the latent factors with a sequential Adam-adjusting hyper-parameters PSO algorithm. First, we design the Adam incremental vector for a particle and construct the Adam-enhanced evolution process for particles. Second, we refine all the latent factors of the target matrix sequentially with our proposed Adam-enhanced PSO's process. The experimental results on four real datasets demonstrate that our proposed model achieves higher prediction accuracy with its peers.
A Dynamic-Neighbor Particle Swarm Optimizer for Accurate Latent Factor Analysis
Feb 23, 2023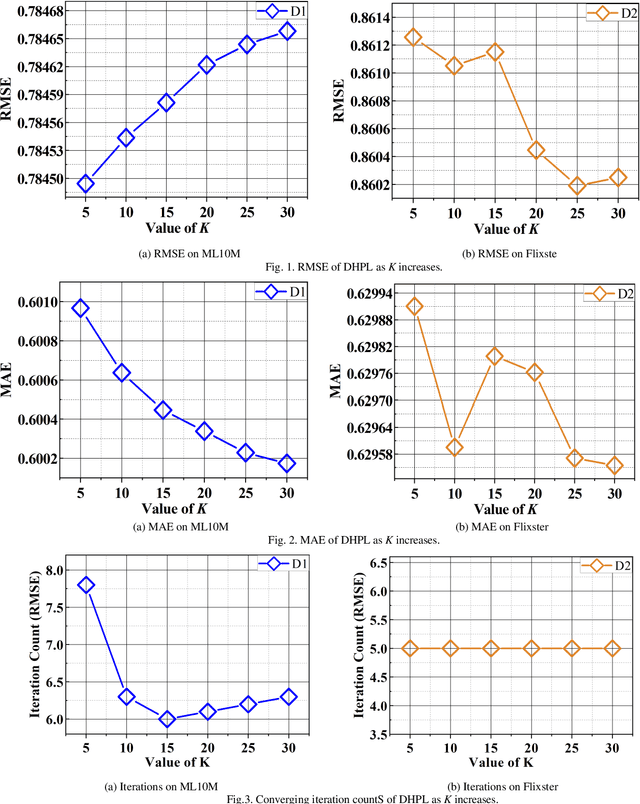
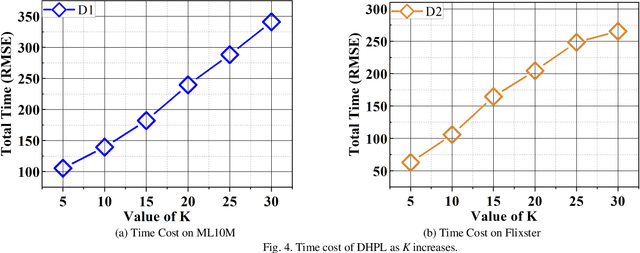

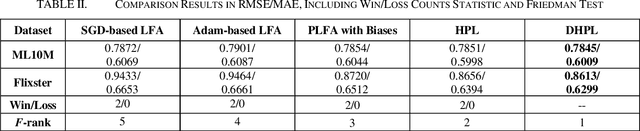
High-Dimensional and Incomplete matrices, which usually contain a large amount of valuable latent information, can be well represented by a Latent Factor Analysis model. The performance of an LFA model heavily rely on its optimization process. Thereby, some prior studies employ the Particle Swarm Optimization to enhance an LFA model's optimization process. However, the particles within the swarm follow the static evolution paths and only share the global best information, which limits the particles' searching area to cause sub-optimum issue. To address this issue, this paper proposes a Dynamic-neighbor-cooperated Hierarchical PSO-enhanced LFA model with two-fold main ideas. First is the neighbor-cooperated strategy, which enhances the randomly chosen neighbor's velocity for particles' evolution. Second is the dynamic hyper-parameter tunning. Extensive experiments on two benchmark datasets are conducted to evaluate the proposed DHPL model. The results substantiate that DHPL achieves a higher accuracy without hyper-parameters tunning than the existing PSO-incorporated LFA models in representing an HDI matrix.
BALanCe: Deep Bayesian Active Learning via Equivalence Class Annealing
Dec 27, 2021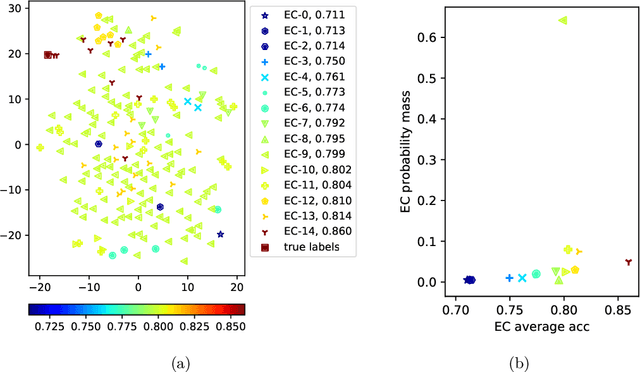


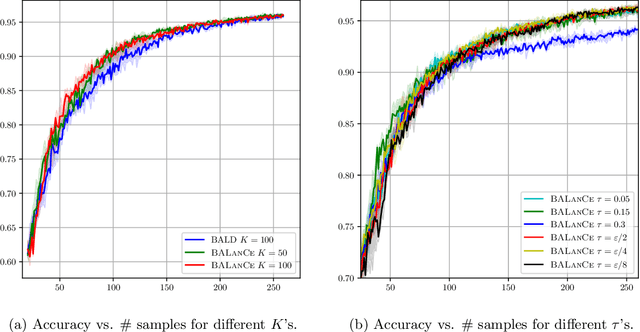
Active learning has demonstrated data efficiency in many fields. Existing active learning algorithms, especially in the context of deep Bayesian active models, rely heavily on the quality of uncertainty estimations of the model. However, such uncertainty estimates could be heavily biased, especially with limited and imbalanced training data. In this paper, we propose BALanCe, a Bayesian deep active learning framework that mitigates the effect of such biases. Concretely, BALanCe employs a novel acquisition function which leverages the structure captured by equivalence hypothesis classes and facilitates differentiation among different equivalence classes. Intuitively, each equivalence class consists of instantiations of deep models with similar predictions, and BALanCe adaptively adjusts the size of the equivalence classes as learning progresses. Besides the fully sequential setting, we further propose Batch-BALanCe -- a generalization of the sequential algorithm to the batched setting -- to efficiently select batches of training examples that are jointly effective for model improvement. We show that Batch-BALanCe achieves state-of-the-art performance on several benchmark datasets for active learning, and that both algorithms can effectively handle realistic challenges that often involve multi-class and imbalanced data.
Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model
Nov 27, 2016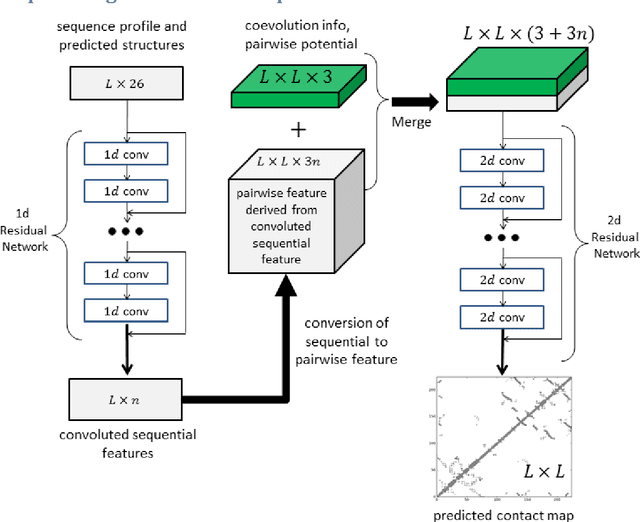
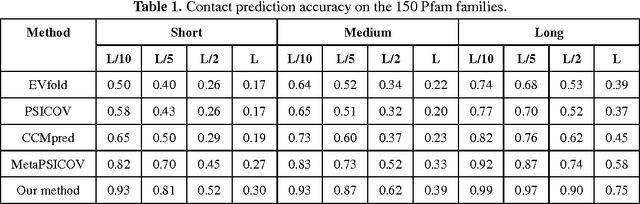
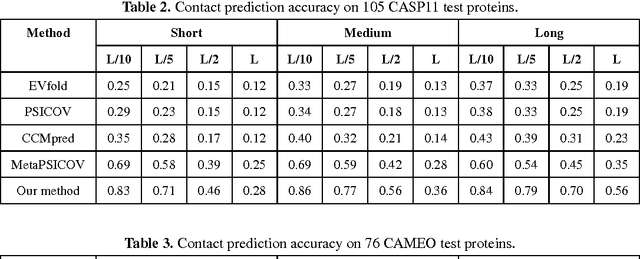
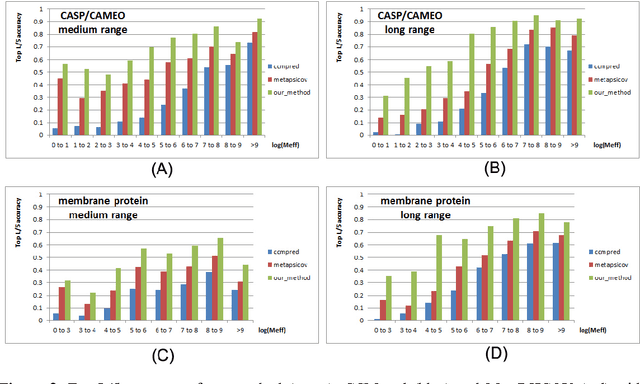
Recently exciting progress has been made on protein contact prediction, but the predicted contacts for proteins without many sequence homologs is still of low quality and not very useful for de novo structure prediction. This paper presents a new deep learning method that predicts contacts by integrating both evolutionary coupling (EC) and sequence conservation information through an ultra-deep neural network formed by two deep residual networks. This deep neural network allows us to model very complex sequence-contact relationship as well as long-range inter-contact correlation. Our method greatly outperforms existing contact prediction methods and leads to much more accurate contact-assisted protein folding. Tested on three datasets of 579 proteins, the average top L long-range prediction accuracy obtained our method, the representative EC method CCMpred and the CASP11 winner MetaPSICOV is 0.47, 0.21 and 0.30, respectively; the average top L/10 long-range accuracy of our method, CCMpred and MetaPSICOV is 0.77, 0.47 and 0.59, respectively. Ab initio folding using our predicted contacts as restraints can yield correct folds (i.e., TMscore>0.6) for 203 test proteins, while that using MetaPSICOV- and CCMpred-predicted contacts can do so for only 79 and 62 proteins, respectively. Further, our contact-assisted models have much better quality than template-based models. Using our predicted contacts as restraints, we can (ab initio) fold 208 of the 398 membrane proteins with TMscore>0.5. By contrast, when the training proteins of our method are used as templates, homology modeling can only do so for 10 of them. One interesting finding is that even if we do not train our prediction models with any membrane proteins, our method works very well on membrane protein prediction. Finally, in recent blind CAMEO benchmark our method successfully folded 5 test proteins with a novel fold.