 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient and generalizable prediction of molecular alterations in multiple cancer cohorts using H&E whole slide images
Jul 22, 2024Molecular testing of tumor samples for targetable biomarkers is restricted by a lack of standardization, turnaround-time, cost, and tissue availability across cancer types. Additionally, targetable alterations of low prevalence may not be tested in routine workflows. Algorithms that predict DNA alterations from routinely generated hematoxylin and eosin (H&E)-stained images could prioritize samples for confirmatory molecular testing. Costs and the necessity of a large number of samples containing mutations limit approaches that train individual algorithms for each alteration. In this work, models were trained for simultaneous prediction of multiple DNA alterations from H&E images using a multi-task approach. Compared to biomarker-specific models, this approach performed better on average, with pronounced gains for rare mutations. The models reasonably generalized to independent temporal-holdout, externally-stained, and multi-site TCGA test sets. Additionally, whole slide image embeddings derived using multi-task models demonstrated strong performance in downstream tasks that were not a part of training. Overall, this is a promising approach to develop clinically useful algorithms that provide multiple actionable predictions from a single slide.
Large Language Models with Retrieval-Augmented Generation for Zero-Shot Disease Phenotyping
Dec 11, 2023Identifying disease phenotypes from electronic health records (EHRs) is critical for numerous secondary uses. Manually encoding physician knowledge into rules is particularly challenging for rare diseases due to inadequate EHR coding, necessitating review of clinical notes. Large language models (LLMs) offer promise in text understanding but may not efficiently handle real-world clinical documentation. We propose a zero-shot LLM-based method enriched by retrieval-augmented generation and MapReduce, which pre-identifies disease-related text snippets to be used in parallel as queries for the LLM to establish diagnosis. We show that this method as applied to pulmonary hypertension (PH), a rare disease characterized by elevated arterial pressures in the lungs, significantly outperforms physician logic rules ($F_1$ score of 0.62 vs. 0.75). This method has the potential to enhance rare disease cohort identification, expanding the scope of robust clinical research and care gap identification.
Development and Validation of a Deep Learning-Based Microsatellite Instability Predictor from Prostate Cancer Whole-Slide Images
Oct 12, 2023Microsatellite instability-high (MSI-H) is a tumor agnostic biomarker for immune checkpoint inhibitor therapy. However, MSI status is not routinely tested in prostate cancer, in part due to low prevalence and assay cost. As such, prediction of MSI status from hematoxylin and eosin (H&E) stained whole-slide images (WSIs) could identify prostate cancer patients most likely to benefit from confirmatory testing and becoming eligible for immunotherapy. Prostate biopsies and surgical resections from de-identified records of consecutive prostate cancer patients referred to our institution were analyzed. Their MSI status was determined by next generation sequencing. Patients before a cutoff date were split into an algorithm development set (n=4015, MSI-H 1.8%) and a paired validation set (n=173, MSI-H 19.7%) that consisted of two serial sections from each sample, one stained and scanned internally and the other at an external site. Patients after the cutoff date formed the temporal validation set (n=1350, MSI-H 2.3%). Attention-based multiple instance learning models were trained to predict MSI-H from H&E WSIs. The MSI-H predictor achieved area under the receiver operating characteristic curve values of 0.78 (95% CI [0.69-0.86]), 0.72 (95% CI [0.63-0.81]), and 0.72 (95% CI [0.62-0.82]) on the internally prepared, externally prepared, and temporal validation sets, respectively. While MSI-H status is significantly correlated with Gleason score, the model remained predictive within each Gleason score subgroup. In summary, we developed and validated an AI-based MSI-H diagnostic model on a large real-world cohort of routine H&E slides, which effectively generalized to externally stained and scanned samples and a temporally independent validation cohort. This algorithm has the potential to direct prostate cancer patients toward immunotherapy and to identify MSI-H cases secondary to Lynch syndrome.
AI-augmented histopathologic review using image analysis to optimize DNA yield and tumor purity from FFPE slides
Apr 07, 2022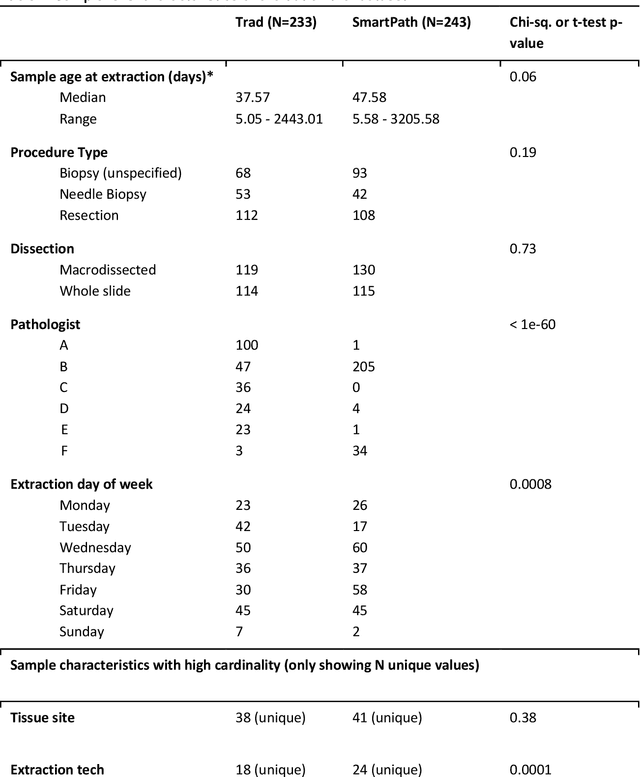

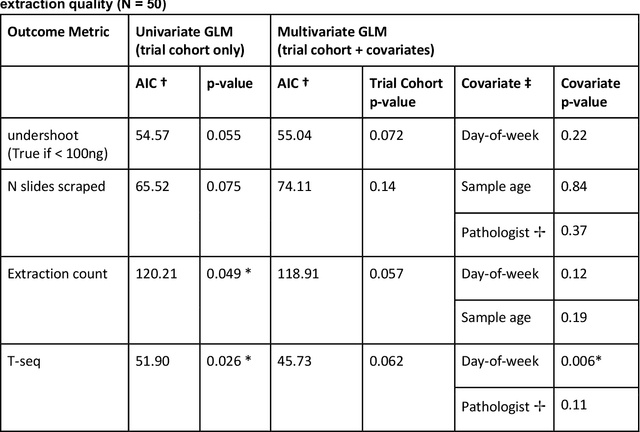

To achieve minimum DNA input and tumor purity requirements for next-generation sequencing (NGS), pathologists visually estimate macrodissection and slide count decisions. Misestimation may cause tissue waste and increased laboratory costs. We developed an AI-augmented smart pathology review system (SmartPath) to empower pathologists with quantitative metrics for determining tissue extraction parameters. Using digitized H&E-stained FFPE slides as inputs, SmartPath segments tumors, extracts cell-based features, and suggests macrodissection areas. To predict DNA yield per slide, the extracted features are correlated with known DNA yields. Then, a pathologist-defined target yield divided by the predicted DNA yield/slide gives the number of slides to scrape. Following model development, an internal validation trial was conducted within the Tempus Labs molecular sequencing laboratory. We evaluated our system on 501 clinical colorectal cancer slides, where half received SmartPath-augmented review and half traditional pathologist review. The SmartPath cohort had 25% more DNA yields within a desired target range of 100-2000ng. The SmartPath system recommended fewer slides to scrape for large tissue sections, saving tissue in these cases. Conversely, SmartPath recommended more slides to scrape for samples with scant tissue sections, helping prevent costly re-extraction due to insufficient extraction yield. A statistical analysis was performed to measure the impact of covariates on the results, offering insights on how to improve future applications of SmartPath. Overall, the study demonstrated that AI-augmented histopathologic review using SmartPath could decrease tissue waste, sequencing time, and laboratory costs by optimizing DNA yields and tumor purity.
Imaging-based histological features are predictive of MET alterations in Non-Small Cell Lung Cancer
Mar 29, 2022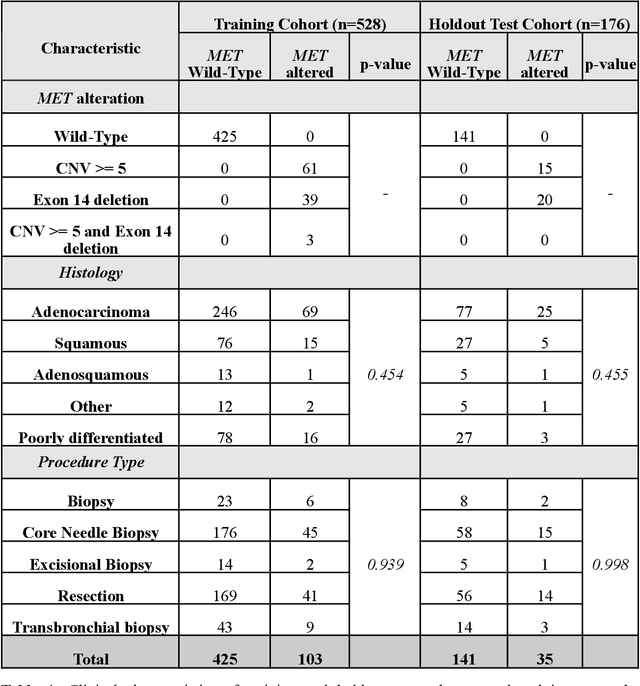
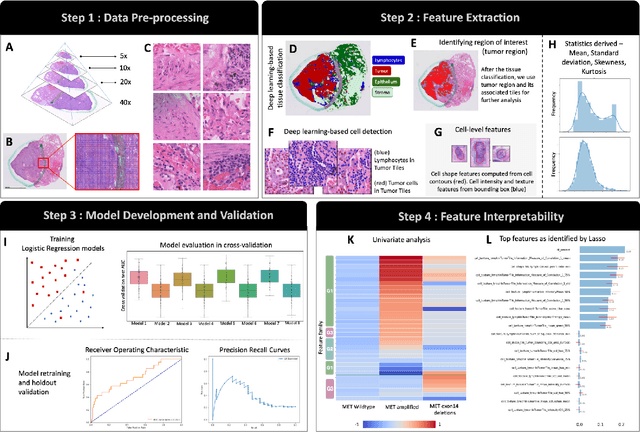
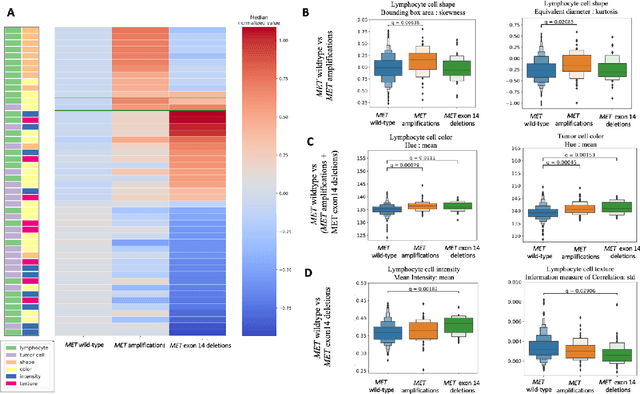
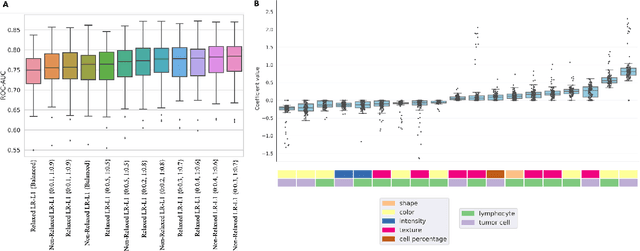
MET is a proto-oncogene whose somatic activation in non-small cell lung cancer leads to increased cell growth and tumor progression. The two major classes of MET alterations are gene amplification and exon 14 deletion, both of which are therapeutic targets and detectable using existing molecular assays. However, existing tests are limited by their consumption of valuable tissue, cost and complexity that prevent widespread use. MET alterations could have an effect on cell morphology, and quantifying these associations could open new avenues for research and development of morphology-based screening tools. Using H&E-stained whole slide images (WSIs), we investigated the association of distinct cell-morphological features with MET amplifications and MET exon 14 deletions. We found that cell shape, color, grayscale intensity and texture-based features from both tumor infiltrating lymphocytes and tumor cells distinguished MET wild-type from MET amplified or MET exon 14 deletion cases. The association of individual cell features with MET alterations suggested a predictive model could distinguish MET wild-type from MET amplification or MET exon 14 deletion. We therefore developed an L1-penalized logistic regression model, achieving a mean Area Under the Receiver Operating Characteristic Curve (ROC-AUC) of 0.77 +/- 0.05sd in cross-validation and 0.77 on an independent holdout test set. A sparse set of 43 features differentiated these classes, which included features similar to what was found in the univariate analysis as well as the percent of tumor cells in the tissue. Our study demonstrates that MET alterations result in a detectable morphological signal in tumor cells and lymphocytes. These results suggest that development of low-cost predictive models based on H&E-stained WSIs may improve screening for MET altered tumors.
Deep Orthogonal Fusion: Multimodal Prognostic Biomarker Discovery Integrating Radiology, Pathology, Genomic, and Clinical Data
Jul 01, 2021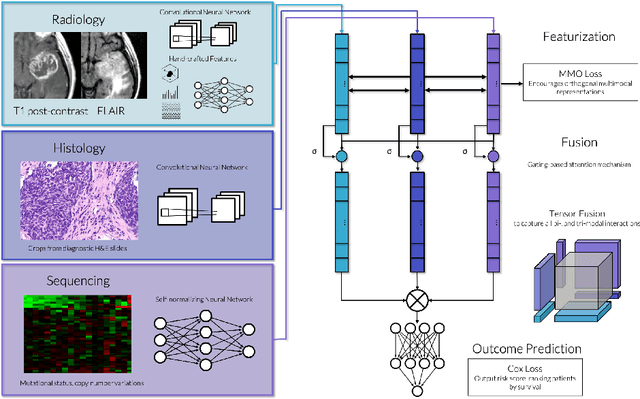
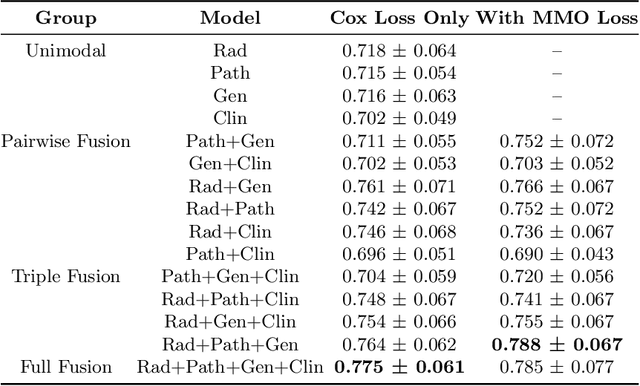
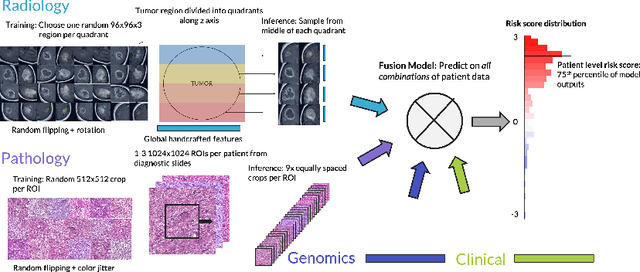
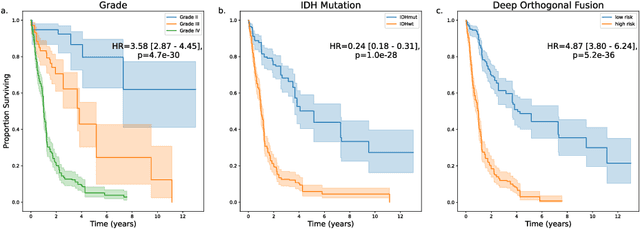
Clinical decision-making in oncology involves multimodal data such as radiology scans, molecular profiling, histopathology slides, and clinical factors. Despite the importance of these modalities individually, no deep learning framework to date has combined them all to predict patient prognosis. Here, we predict the overall survival (OS) of glioma patients from diverse multimodal data with a Deep Orthogonal Fusion (DOF) model. The model learns to combine information from multiparametric MRI exams, biopsy-based modalities (such as H&E slide images and/or DNA sequencing), and clinical variables into a comprehensive multimodal risk score. Prognostic embeddings from each modality are learned and combined via attention-gated tensor fusion. To maximize the information gleaned from each modality, we introduce a multimodal orthogonalization (MMO) loss term that increases model performance by incentivizing constituent embeddings to be more complementary. DOF predicts OS in glioma patients with a median C-index of 0.788 +/- 0.067, significantly outperforming (p=0.023) the best performing unimodal model with a median C-index of 0.718 +/- 0.064. The prognostic model significantly stratifies glioma patients by OS within clinical subsets, adding further granularity to prognostic clinical grading and molecular subtyping.
Predicting Prostate Cancer-Specific Mortality with A.I.-based Gleason Grading
Nov 25, 2020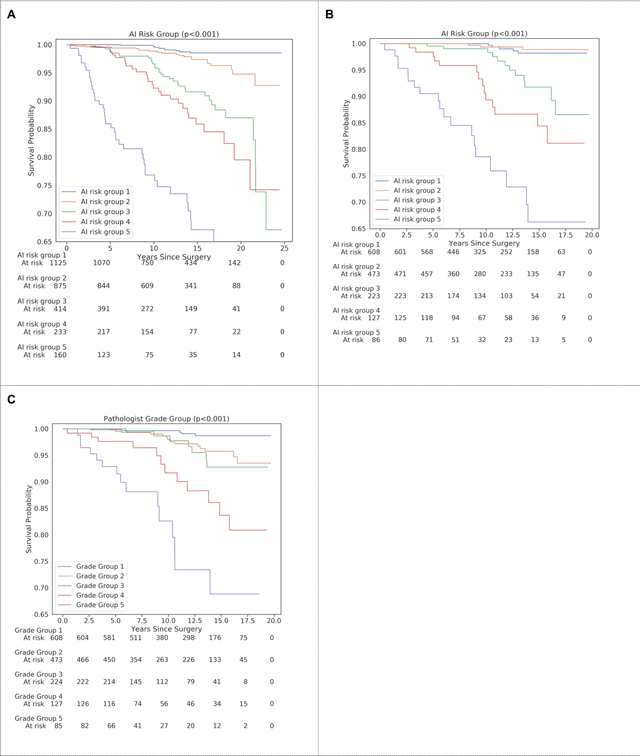
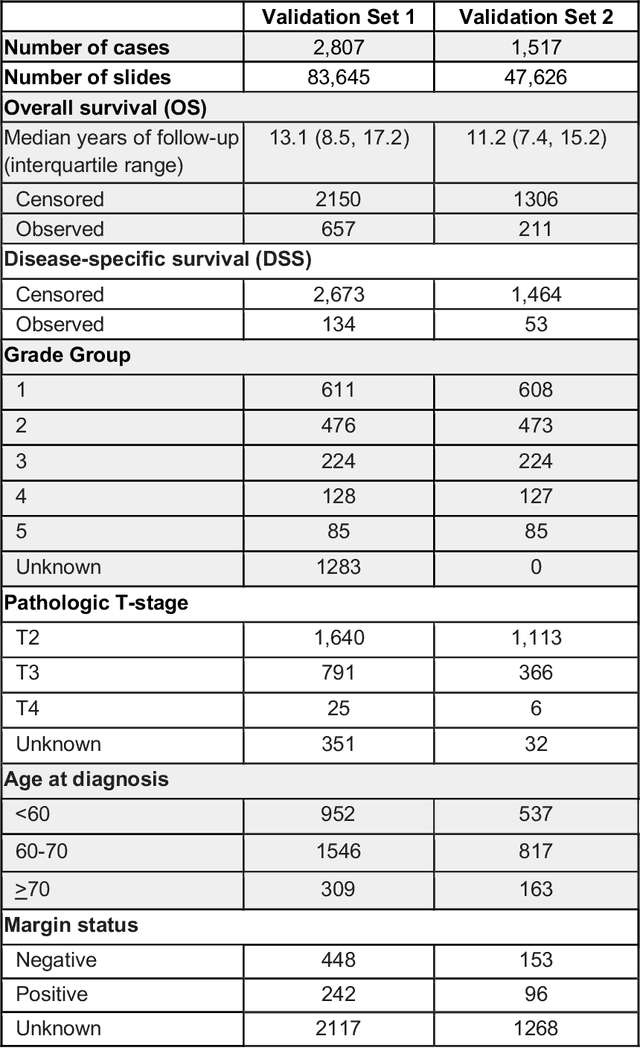
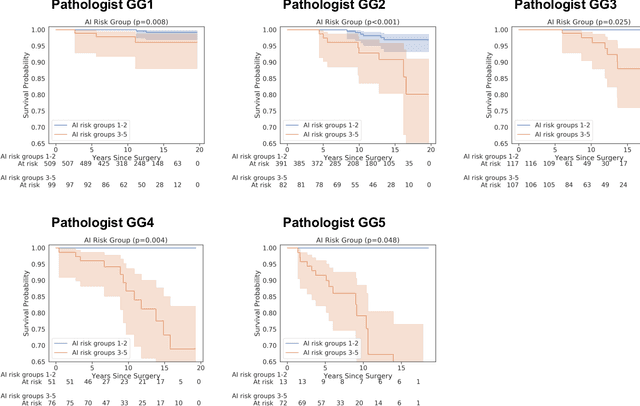
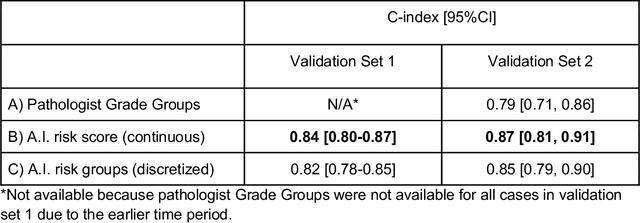
Gleason grading of prostate cancer is an important prognostic factor but suffers from poor reproducibility, particularly among non-subspecialist pathologists. Although artificial intelligence (A.I.) tools have demonstrated Gleason grading on-par with expert pathologists, it remains an open question whether A.I. grading translates to better prognostication. In this study, we developed a system to predict prostate-cancer specific mortality via A.I.-based Gleason grading and subsequently evaluated its ability to risk-stratify patients on an independent retrospective cohort of 2,807 prostatectomy cases from a single European center with 5-25 years of follow-up (median: 13, interquartile range 9-17). The A.I.'s risk scores produced a C-index of 0.84 (95%CI 0.80-0.87) for prostate cancer-specific mortality. Upon discretizing these risk scores into risk groups analogous to pathologist Grade Groups (GG), the A.I. had a C-index of 0.82 (95%CI 0.78-0.85). On the subset of cases with a GG in the original pathology report (n=1,517), the A.I.'s C-indices were 0.87 and 0.85 for continuous and discrete grading, respectively, compared to 0.79 (95%CI 0.71-0.86) for GG obtained from the reports. These represent improvements of 0.08 (95%CI 0.01-0.15) and 0.07 (95%CI 0.00-0.14) respectively. Our results suggest that A.I.-based Gleason grading can lead to effective risk-stratification and warrants further evaluation for improving disease management.
Interpretable Survival Prediction for Colorectal Cancer using Deep Learning
Nov 17, 2020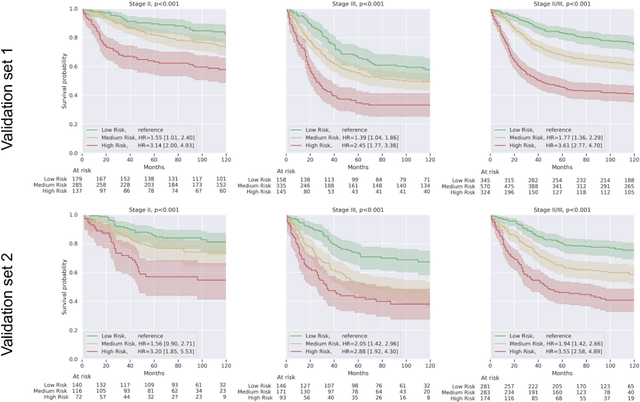
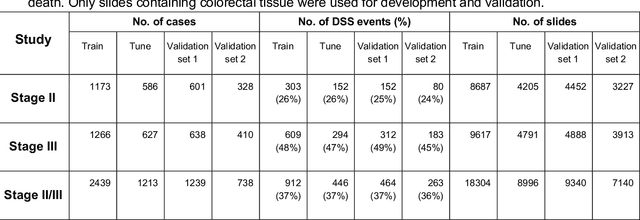
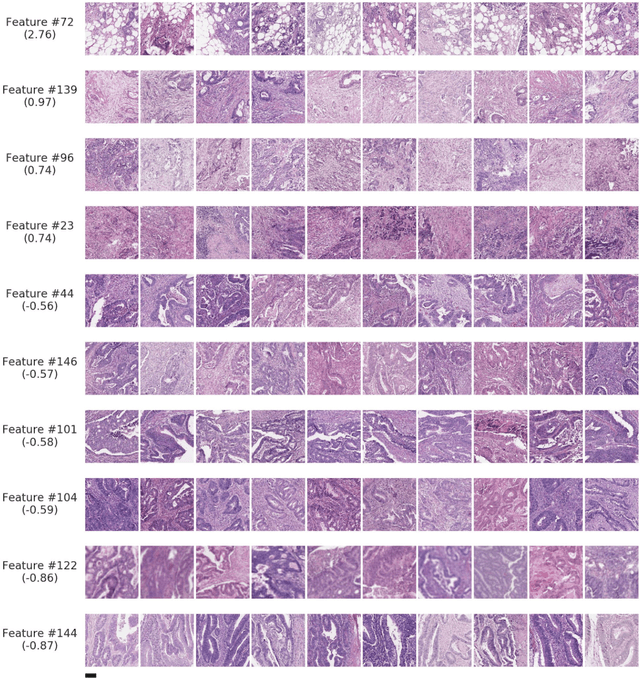
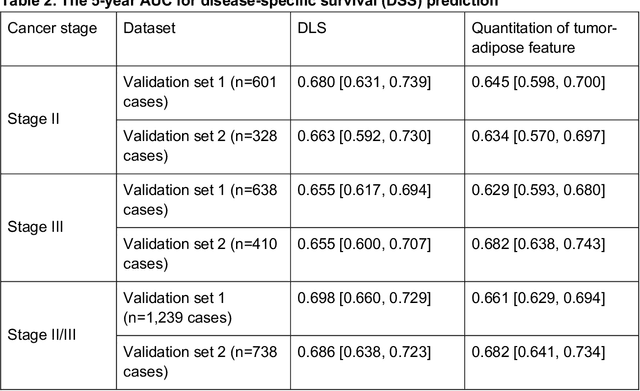
Deriving interpretable prognostic features from deep-learning-based prognostic histopathology models remains a challenge. In this study, we developed a deep learning system (DLS) for predicting disease specific survival for stage II and III colorectal cancer using 3,652 cases (27,300 slides). When evaluated on two validation datasets containing 1,239 cases (9,340 slides) and 738 cases (7,140 slides) respectively, the DLS achieved a 5-year disease-specific survival AUC of 0.70 (95%CI 0.66-0.73) and 0.69 (95%CI 0.64-0.72), and added significant predictive value to a set of 9 clinicopathologic features. To interpret the DLS, we explored the ability of different human-interpretable features to explain the variance in DLS scores. We observed that clinicopathologic features such as T-category, N-category, and grade explained a small fraction of the variance in DLS scores (R2=18% in both validation sets). Next, we generated human-interpretable histologic features by clustering embeddings from a deep-learning based image-similarity model and showed that they explain the majority of the variance (R2 of 73% to 80%). Furthermore, the clustering-derived feature most strongly associated with high DLS scores was also highly prognostic in isolation. With a distinct visual appearance (poorly differentiated tumor cell clusters adjacent to adipose tissue), this feature was identified by annotators with 87.0-95.5% accuracy. Our approach can be used to explain predictions from a prognostic deep learning model and uncover potentially-novel prognostic features that can be reliably identified by people for future validation studies.
Deep learning-based survival prediction for multiple cancer types using histopathology images
Dec 16, 2019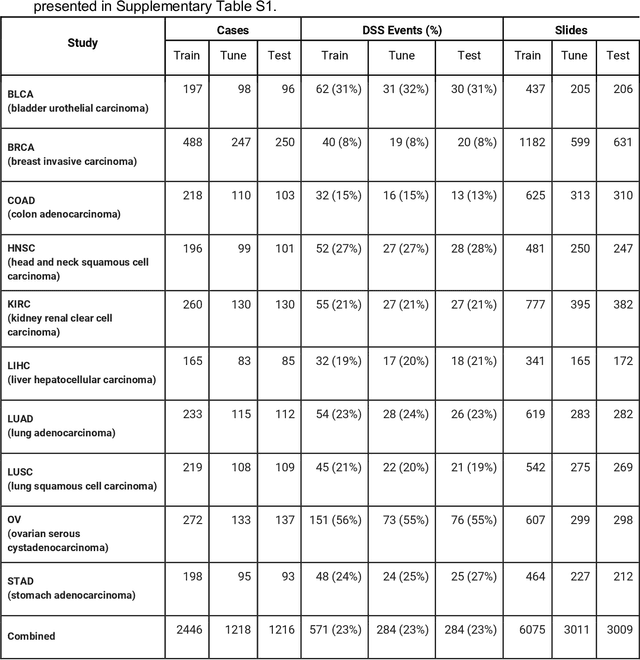
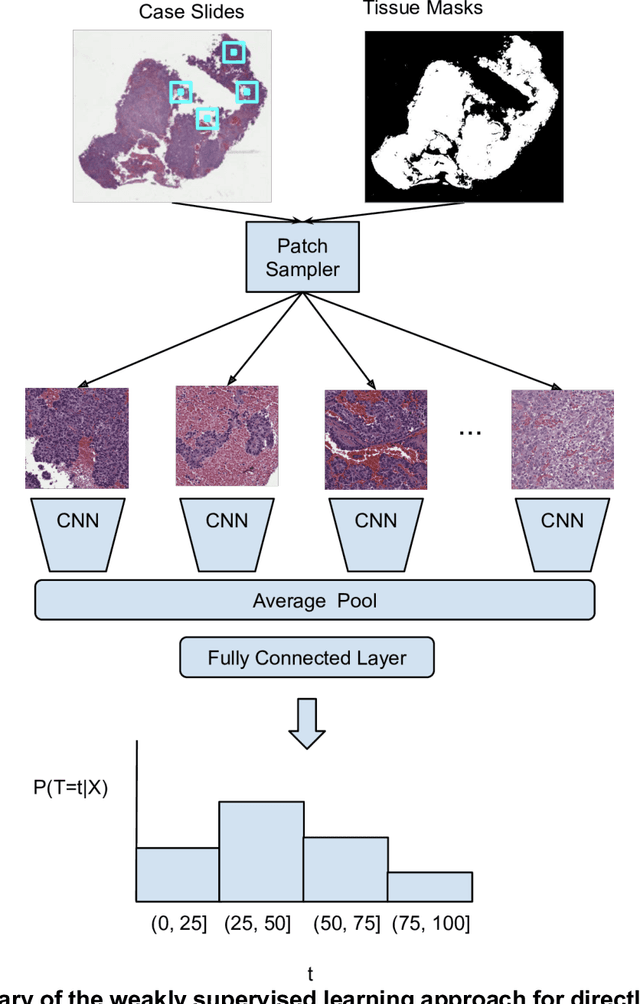
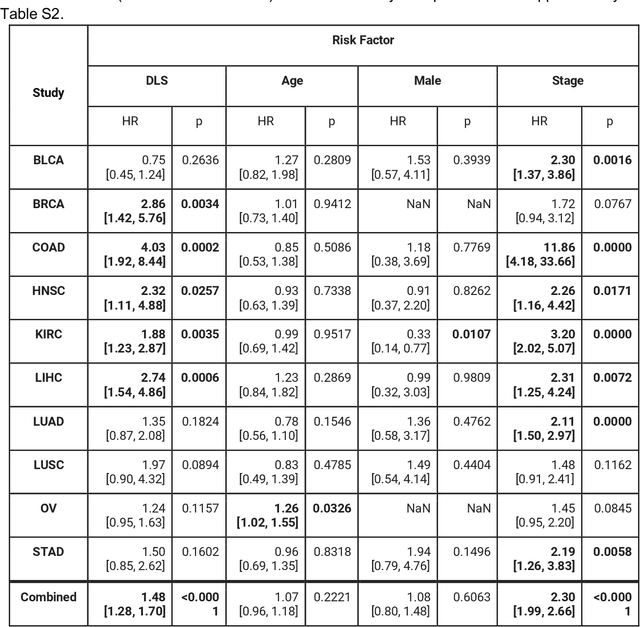
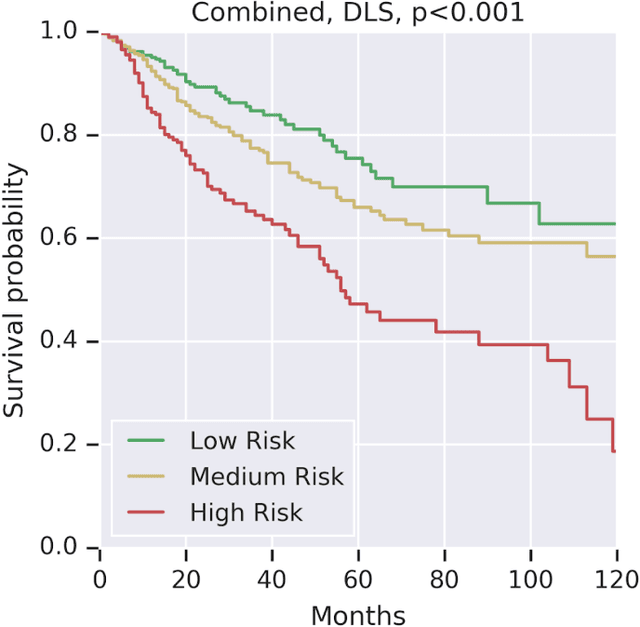
Prognostic information at diagnosis has important implications for cancer treatment and monitoring. Although cancer staging, histopathological assessment, molecular features, and clinical variables can provide useful prognostic insights, improving risk stratification remains an active research area. We developed a deep learning system (DLS) to predict disease specific survival across 10 cancer types from The Cancer Genome Atlas (TCGA). We used a weakly-supervised approach without pixel-level annotations, and tested three different survival loss functions. The DLS was developed using 9,086 slides from 3,664 cases and evaluated using 3,009 slides from 1,216 cases. In multivariable Cox regression analysis of the combined cohort including all 10 cancers, the DLS was significantly associated with disease specific survival (hazard ratio of 1.58, 95% CI 1.28-1.70, p<0.0001) after adjusting for cancer type, stage, age, and sex. In a per-cancer adjusted subanalysis, the DLS remained a significant predictor of survival in 5 of 10 cancer types. Compared to a baseline model including stage, age, and sex, the c-index of the model demonstrated an absolute 3.7% improvement (95% CI 1.0-6.5) in the combined cohort. Additionally, our models stratified patients within individual cancer stages, particularly stage II (p=0.025) and stage III (p<0.001). By developing and evaluating prognostic models across multiple cancer types, this work represents one of the most comprehensive studies exploring the direct prediction of clinical outcomes using deep learning and histopathology images. Our analysis demonstrates the potential for this approach to provide prognostic information in multiple cancer types, and even within specific pathologic stages. However, given the relatively small number of clinical events, we observed wide confidence intervals, suggesting that future work will benefit from larger datasets.
Similar Image Search for Histopathology: SMILY
Feb 06, 2019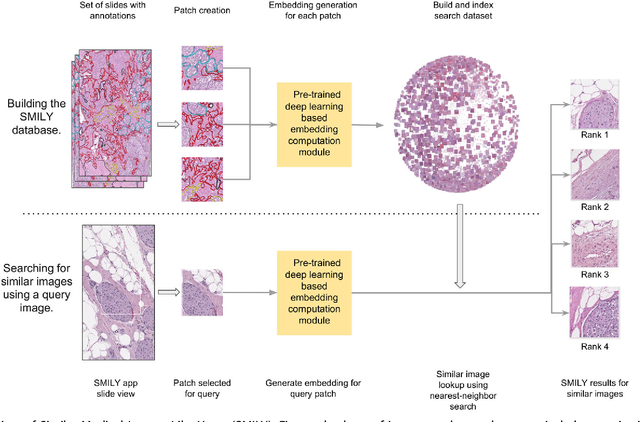
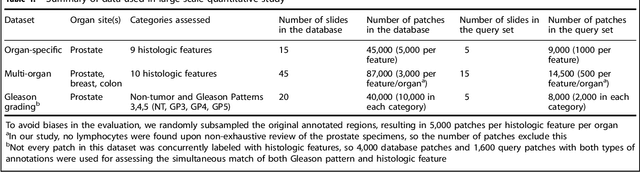
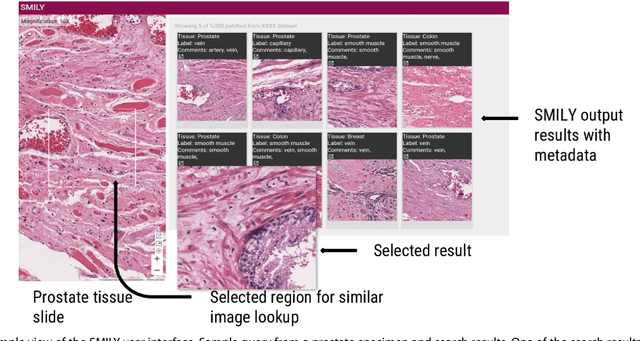
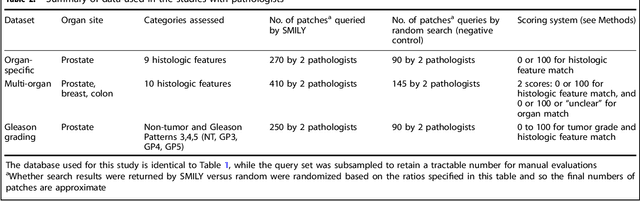
The increasing availability of large institutional and public histopathology image datasets is enabling the searching of these datasets for diagnosis, research, and education. Though these datasets typically have associated metadata such as diagnosis or clinical notes, even carefully curated datasets rarely contain annotations of the location of regions of interest on each image. Because pathology images are extremely large (up to 100,000 pixels in each dimension), further laborious visual search of each image may be needed to find the feature of interest. In this paper, we introduce a deep learning based reverse image search tool for histopathology images: Similar Medical Images Like Yours (SMILY). We assessed SMILY's ability to retrieve search results in two ways: using pathologist-provided annotations, and via prospective studies where pathologists evaluated the quality of SMILY search results. As a negative control in the second evaluation, pathologists were blinded to whether search results were retrieved by SMILY or randomly. In both types of assessments, SMILY was able to retrieve search results with similar histologic features, organ site, and prostate cancer Gleason grade compared with the original query. SMILY may be a useful general-purpose tool in the pathologist's arsenal, to improve the efficiency of searching large archives of histopathology images, without the need to develop and implement specific tools for each application.