 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrediction of MET Overexpression in Non-Small Cell Lung Adenocarcinomas from Hematoxylin and Eosin Images
Oct 12, 2023MET protein overexpression is a targetable event in non-small cell lung cancer (NSCLC) and is the subject of active drug development. Challenges in identifying patients for these therapies include lack of access to validated testing, such as standardized immunohistochemistry (IHC) assessment, and consumption of valuable tissue for a single gene/protein assay. Development of pre-screening algorithms using routinely available digitized hematoxylin and eosin (H&E)-stained slides to predict MET overexpression could promote testing for those who will benefit most. While assessment of MET expression using IHC is currently not routinely performed in NSCLC, next-generation sequencing is common and in some cases includes RNA expression panel testing. In this work, we leveraged a large database of matched H&E slides and RNA expression data to train a weakly supervised model to predict MET RNA overexpression directly from H&E images. This model was evaluated on an independent holdout test set of 300 over-expressed and 289 normal patients, demonstrating an ROC-AUC of 0.70 (95th percentile interval: 0.66 - 0.74) with stable performance characteristics across different patient clinical variables and robust to synthetic noise on the test set. These results suggest that H&E-based predictive models could be useful to prioritize patients for confirmatory testing of MET protein or MET gene expression status.
Imaging-based histological features are predictive of MET alterations in Non-Small Cell Lung Cancer
Mar 29, 2022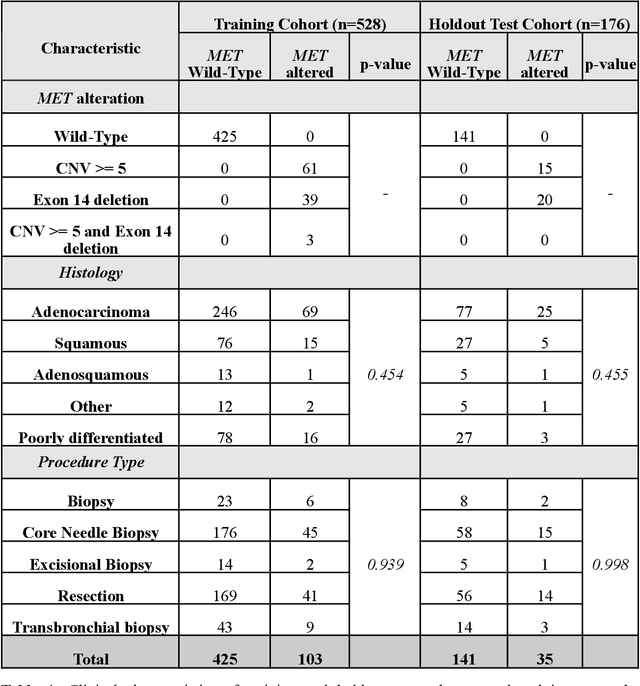
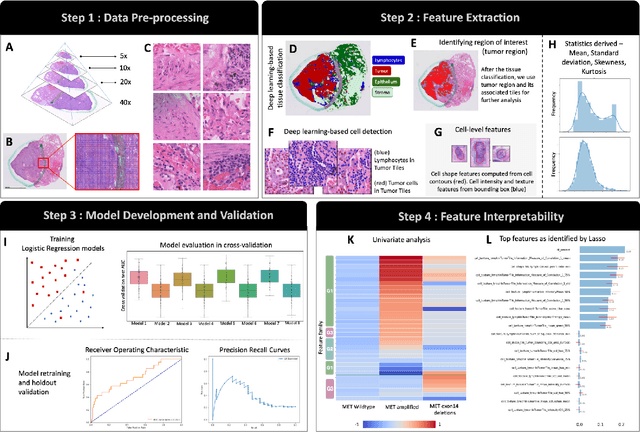
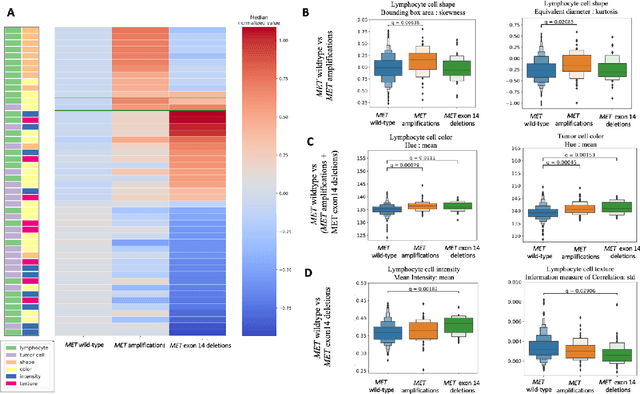
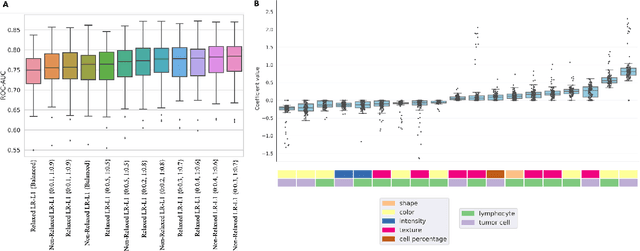
MET is a proto-oncogene whose somatic activation in non-small cell lung cancer leads to increased cell growth and tumor progression. The two major classes of MET alterations are gene amplification and exon 14 deletion, both of which are therapeutic targets and detectable using existing molecular assays. However, existing tests are limited by their consumption of valuable tissue, cost and complexity that prevent widespread use. MET alterations could have an effect on cell morphology, and quantifying these associations could open new avenues for research and development of morphology-based screening tools. Using H&E-stained whole slide images (WSIs), we investigated the association of distinct cell-morphological features with MET amplifications and MET exon 14 deletions. We found that cell shape, color, grayscale intensity and texture-based features from both tumor infiltrating lymphocytes and tumor cells distinguished MET wild-type from MET amplified or MET exon 14 deletion cases. The association of individual cell features with MET alterations suggested a predictive model could distinguish MET wild-type from MET amplification or MET exon 14 deletion. We therefore developed an L1-penalized logistic regression model, achieving a mean Area Under the Receiver Operating Characteristic Curve (ROC-AUC) of 0.77 +/- 0.05sd in cross-validation and 0.77 on an independent holdout test set. A sparse set of 43 features differentiated these classes, which included features similar to what was found in the univariate analysis as well as the percent of tumor cells in the tissue. Our study demonstrates that MET alterations result in a detectable morphological signal in tumor cells and lymphocytes. These results suggest that development of low-cost predictive models based on H&E-stained WSIs may improve screening for MET altered tumors.