 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHistopathology Based AI Model Predicts Anti-Angiogenic Therapy Response in Renal Cancer Clinical Trial
May 28, 2024



Predictive biomarkers of treatment response are lacking for metastatic clear cell renal cell carcinoma (ccRCC), a tumor type that is treated with angiogenesis inhibitors, immune checkpoint inhibitors, mTOR inhibitors and a HIF2 inhibitor. The Angioscore, an RNA-based quantification of angiogenesis, is arguably the best candidate to predict anti-angiogenic (AA) response. However, the clinical adoption of transcriptomic assays faces several challenges including standardization, time delay, and high cost. Further, ccRCC tumors are highly heterogenous, and sampling multiple areas for sequencing is impractical. Here we present a novel deep learning (DL) approach to predict the Angioscore from ubiquitous histopathology slides. To overcome the lack of interpretability, one of the biggest limitations of typical DL models, our model produces a visual vascular network which is the basis of the model's prediction. To test its reliability, we applied this model to multiple cohorts including a clinical trial dataset. Our model accurately predicts the RNA-based Angioscore on multiple independent cohorts (spearman correlations of 0.77 and 0.73). Further, the predictions help unravel meaningful biology such as association of angiogenesis with grade, stage, and driver mutation status. Finally, we find our model can predict response to AA therapy, in both a real-world cohort and the IMmotion150 clinical trial. The predictive power of our model vastly exceeds that of CD31, a marker of vasculature, and nearly rivals the performance (c-index 0.66 vs 0.67) of the ground truth RNA-based Angioscore at a fraction of the cost. By providing a robust yet interpretable prediction of the Angioscore from histopathology slides alone, our approach offers insights into angiogenesis biology and AA treatment response.
Imaging-based histological features are predictive of MET alterations in Non-Small Cell Lung Cancer
Mar 29, 2022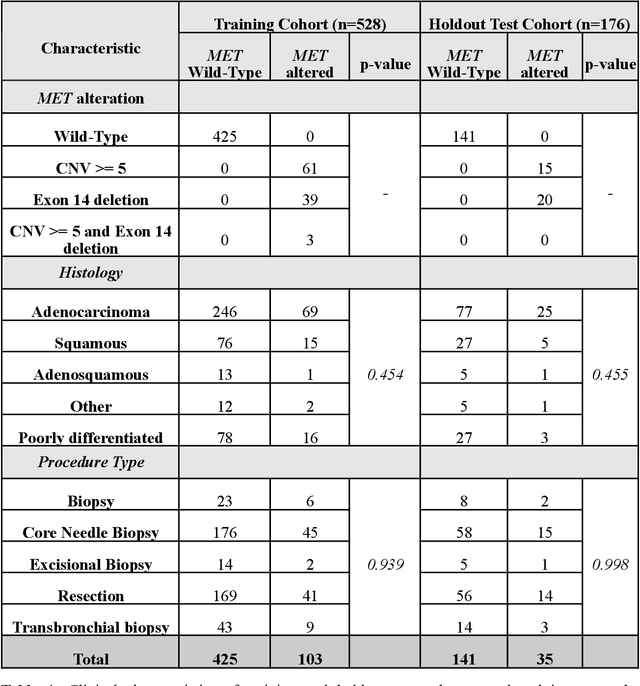
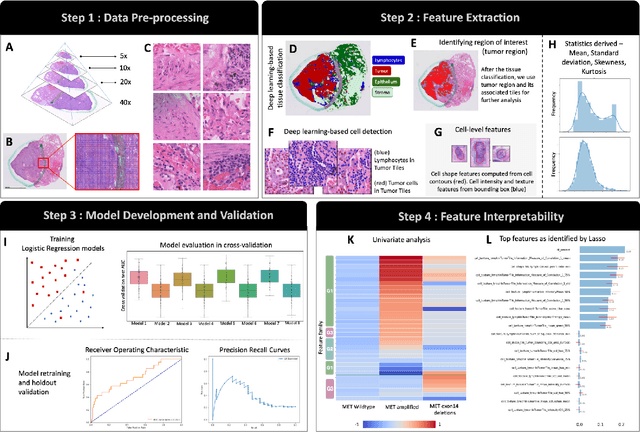
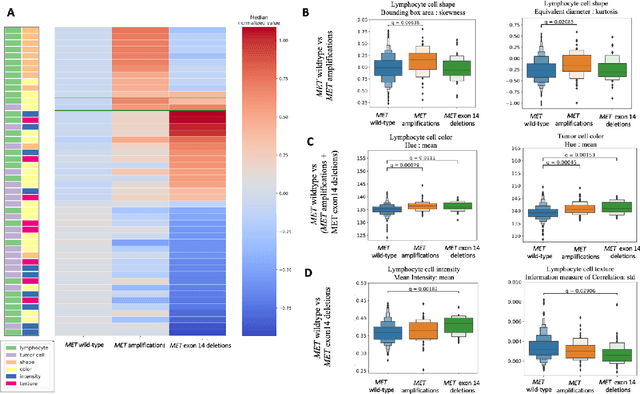
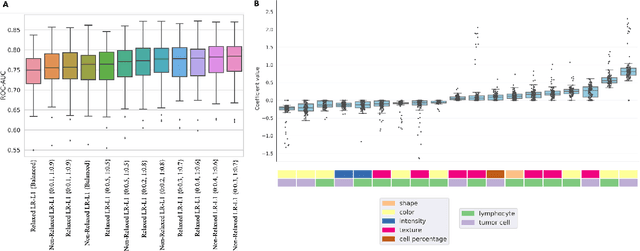
MET is a proto-oncogene whose somatic activation in non-small cell lung cancer leads to increased cell growth and tumor progression. The two major classes of MET alterations are gene amplification and exon 14 deletion, both of which are therapeutic targets and detectable using existing molecular assays. However, existing tests are limited by their consumption of valuable tissue, cost and complexity that prevent widespread use. MET alterations could have an effect on cell morphology, and quantifying these associations could open new avenues for research and development of morphology-based screening tools. Using H&E-stained whole slide images (WSIs), we investigated the association of distinct cell-morphological features with MET amplifications and MET exon 14 deletions. We found that cell shape, color, grayscale intensity and texture-based features from both tumor infiltrating lymphocytes and tumor cells distinguished MET wild-type from MET amplified or MET exon 14 deletion cases. The association of individual cell features with MET alterations suggested a predictive model could distinguish MET wild-type from MET amplification or MET exon 14 deletion. We therefore developed an L1-penalized logistic regression model, achieving a mean Area Under the Receiver Operating Characteristic Curve (ROC-AUC) of 0.77 +/- 0.05sd in cross-validation and 0.77 on an independent holdout test set. A sparse set of 43 features differentiated these classes, which included features similar to what was found in the univariate analysis as well as the percent of tumor cells in the tissue. Our study demonstrates that MET alterations result in a detectable morphological signal in tumor cells and lymphocytes. These results suggest that development of low-cost predictive models based on H&E-stained WSIs may improve screening for MET altered tumors.
Radiomic Deformation and Textural Heterogeneity (R-DepTH) Descriptor to characterize Tumor Field Effect: Application to Survival Prediction in Glioblastoma
Mar 12, 2021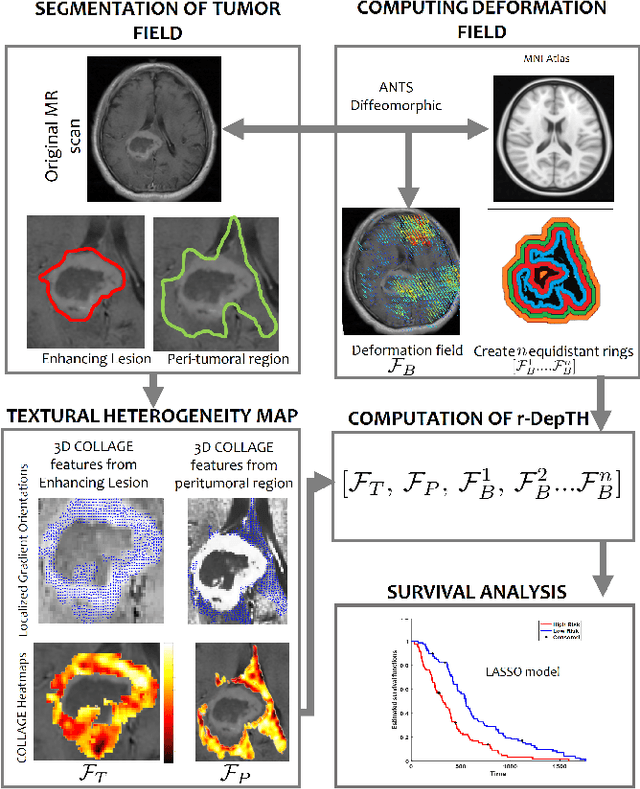
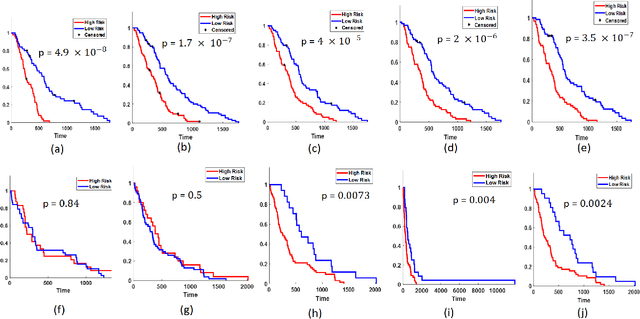
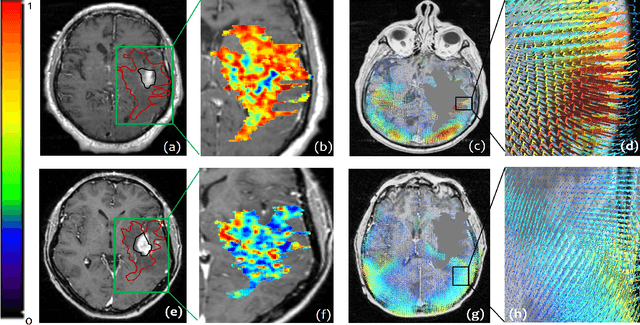
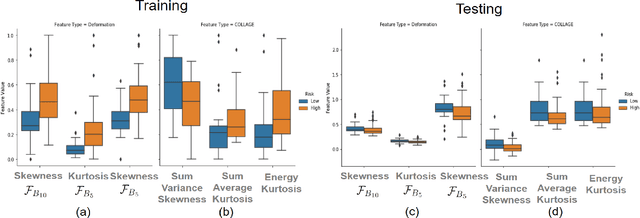
The concept of tumor field effect implies that cancer is a systemic disease with its impact way beyond the visible tumor confines. For instance, in Glioblastoma (GBM), an aggressive brain tumor, the increase in intracranial pressure due to tumor burden often leads to brain herniation and poor outcomes. Our work is based on the rationale that highly aggressive tumors tend to grow uncontrollably, leading to pronounced biomechanical tissue deformations in the normal parenchyma, which when combined with local morphological differences in the tumor confines on MRI scans, will comprehensively capture tumor field effect. Specifically, we present an integrated MRI-based descriptor, radiomic-Deformation and Textural Heterogeneity (r-DepTH). This descriptor comprises measurements of the subtle perturbations in tissue deformations throughout the surrounding normal parenchyma due to mass effect. This involves non-rigidly aligning the patients MRI scans to a healthy atlas via diffeomorphic registration. The resulting inverse mapping is used to obtain the deformation field magnitudes in the normal parenchyma. These measurements are then combined with a 3D texture descriptor, Co-occurrence of Local Anisotropic Gradient Orientations (COLLAGE), which captures the morphological heterogeneity within the tumor confines, on MRI scans. R-DepTH, on N = 207 GBM cases (training set (St) = 128, testing set (Sv) = 79), demonstrated improved prognosis of overall survival by categorizing patients into low- (prolonged survival) and high-risk (poor survival) groups (on St, p-value = 0.0000035, and on Sv, p-value = 0.0024). R-DepTH descriptor may serve as a comprehensive MRI-based prognostic marker of disease aggressiveness and survival in solid tumors.
Spatial-And-Context aware (SpACe) "virtual biopsy" radiogenomic maps to target tumor mutational status on structural MRI
Jun 17, 2020
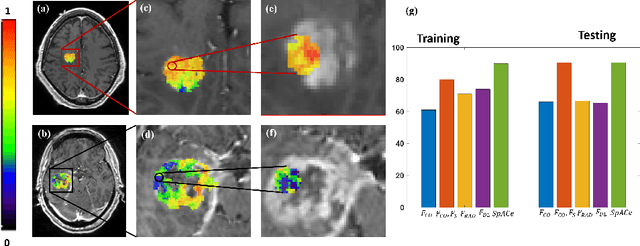
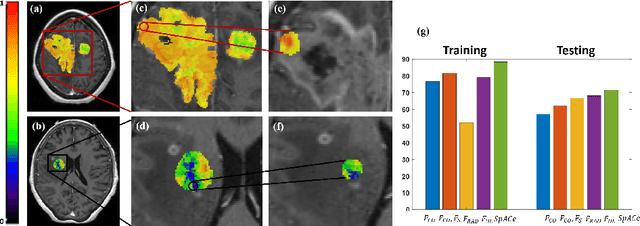
With growing emphasis on personalized cancer-therapies,radiogenomics has shown promise in identifying target tumor mutational status on routine imaging (i.e. MRI) scans. These approaches fall into 2 categories: (1) deep-learning/radiomics (context-based), using image features from the entire tumor to identify the gene mutation status, or (2) atlas (spatial)-based to obtain likelihood of gene mutation status based on population statistics. While many genes (i.e. EGFR, MGMT) are spatially variant, a significant challenge in reliable assessment of gene mutation status on imaging has been the lack of available co-localized ground truth for training the models. We present Spatial-And-Context aware (SpACe) "virtual biopsy" maps that incorporate context-features from co-localized biopsy site along with spatial-priors from population atlases, within a Least Absolute Shrinkage and Selection Operator (LASSO) regression model, to obtain a per-voxel probability of the presence of a mutation status (M+ vs M-). We then use probabilistic pair-wise Markov model to improve the voxel-wise prediction probability. We evaluate the efficacy of SpACe maps on MRI scans with co-localized ground truth obtained from corresponding biopsy, to predict the mutation status of 2 driver genes in Glioblastoma: (1) EGFR (n=91), and (2) MGMT (n=81). When compared against deep-learning (DL) and radiomic models, SpACe maps obtained training and testing accuracies of 90% (n=71) and 90.48% (n=21) in identifying EGFR amplification status,compared to 80% and 71.4% via radiomics, and 74.28% and 65.5% via DL. For MGMT status, training and testing accuracies using SpACe were 88.3% (n=61) and 71.5% (n=20), compared to 52.4% and 66.7% using radiomics,and 79.3% and 68.4% using DL. Following validation,SpACe maps could provide surgical navigation to improve localization of sampling sites for targeting of specific driver genes in cancer.