 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredict Patient Self-reported Race from Skin Histological Images
Jul 30, 2025Artificial Intelligence (AI) has demonstrated success in computational pathology (CPath) for disease detection, biomarker classification, and prognosis prediction. However, its potential to learn unintended demographic biases, particularly those related to social determinants of health, remains understudied. This study investigates whether deep learning models can predict self-reported race from digitized dermatopathology slides and identifies potential morphological shortcuts. Using a multisite dataset with a racially diverse population, we apply an attention-based mechanism to uncover race-associated morphological features. After evaluating three dataset curation strategies to control for confounding factors, the final experiment showed that White and Black demographic groups retained high prediction performance (AUC: 0.799, 0.762), while overall performance dropped to 0.663. Attention analysis revealed the epidermis as a key predictive feature, with significant performance declines when these regions were removed. These findings highlight the need for careful data curation and bias mitigation to ensure equitable AI deployment in pathology. Code available at: https://github.com/sinai-computational-pathology/CPath_SAIF.
A Clinical Benchmark of Public Self-Supervised Pathology Foundation Models
Jul 11, 2024



The use of self-supervised learning (SSL) to train pathology foundation models has increased substantially in the past few years. Notably, several models trained on large quantities of clinical data have been made publicly available in recent months. This will significantly enhance scientific research in computational pathology and help bridge the gap between research and clinical deployment. With the increase in availability of public foundation models of different sizes, trained using different algorithms on different datasets, it becomes important to establish a benchmark to compare the performance of such models on a variety of clinically relevant tasks spanning multiple organs and diseases. In this work, we present a collection of pathology datasets comprising clinical slides associated with clinically relevant endpoints including cancer diagnoses and a variety of biomarkers generated during standard hospital operation from two medical centers. We leverage these datasets to systematically assess the performance of public pathology foundation models and provide insights into best practices for training new foundation models and selecting appropriate pretrained models.
Federated Learning Enables Big Data for Rare Cancer Boundary Detection
Apr 25, 2022Although machine learning (ML) has shown promise in numerous domains, there are concerns about generalizability to out-of-sample data. This is currently addressed by centrally sharing ample, and importantly diverse, data from multiple sites. However, such centralization is challenging to scale (or even not feasible) due to various limitations. Federated ML (FL) provides an alternative to train accurate and generalizable ML models, by only sharing numerical model updates. Here we present findings from the largest FL study to-date, involving data from 71 healthcare institutions across 6 continents, to generate an automatic tumor boundary detector for the rare disease of glioblastoma, utilizing the largest dataset of such patients ever used in the literature (25,256 MRI scans from 6,314 patients). We demonstrate a 33% improvement over a publicly trained model to delineate the surgically targetable tumor, and 23% improvement over the tumor's entire extent. We anticipate our study to: 1) enable more studies in healthcare informed by large and diverse data, ensuring meaningful results for rare diseases and underrepresented populations, 2) facilitate further quantitative analyses for glioblastoma via performance optimization of our consensus model for eventual public release, and 3) demonstrate the effectiveness of FL at such scale and task complexity as a paradigm shift for multi-site collaborations, alleviating the need for data sharing.
Spatial-And-Context aware (SpACe) "virtual biopsy" radiogenomic maps to target tumor mutational status on structural MRI
Jun 17, 2020
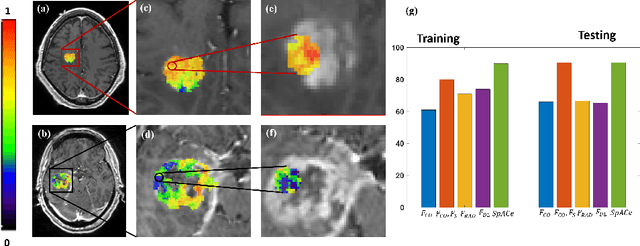
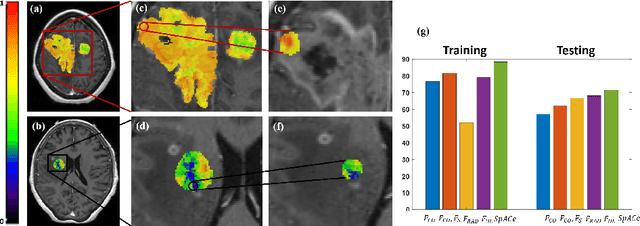
With growing emphasis on personalized cancer-therapies,radiogenomics has shown promise in identifying target tumor mutational status on routine imaging (i.e. MRI) scans. These approaches fall into 2 categories: (1) deep-learning/radiomics (context-based), using image features from the entire tumor to identify the gene mutation status, or (2) atlas (spatial)-based to obtain likelihood of gene mutation status based on population statistics. While many genes (i.e. EGFR, MGMT) are spatially variant, a significant challenge in reliable assessment of gene mutation status on imaging has been the lack of available co-localized ground truth for training the models. We present Spatial-And-Context aware (SpACe) "virtual biopsy" maps that incorporate context-features from co-localized biopsy site along with spatial-priors from population atlases, within a Least Absolute Shrinkage and Selection Operator (LASSO) regression model, to obtain a per-voxel probability of the presence of a mutation status (M+ vs M-). We then use probabilistic pair-wise Markov model to improve the voxel-wise prediction probability. We evaluate the efficacy of SpACe maps on MRI scans with co-localized ground truth obtained from corresponding biopsy, to predict the mutation status of 2 driver genes in Glioblastoma: (1) EGFR (n=91), and (2) MGMT (n=81). When compared against deep-learning (DL) and radiomic models, SpACe maps obtained training and testing accuracies of 90% (n=71) and 90.48% (n=21) in identifying EGFR amplification status,compared to 80% and 71.4% via radiomics, and 74.28% and 65.5% via DL. For MGMT status, training and testing accuracies using SpACe were 88.3% (n=61) and 71.5% (n=20), compared to 52.4% and 66.7% using radiomics,and 79.3% and 68.4% using DL. Following validation,SpACe maps could provide surgical navigation to improve localization of sampling sites for targeting of specific driver genes in cancer.
MRQy: An Open-Source Tool for Quality Control of MR Imaging Data
Apr 13, 2020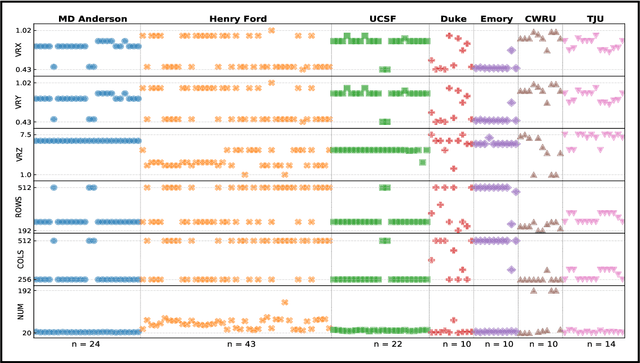

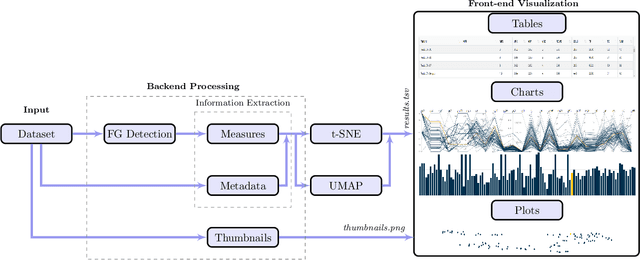
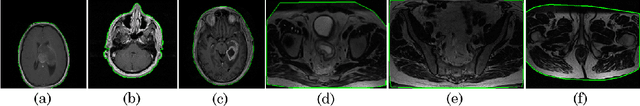
Even as public data repositories such as The Cancer Imaging Archive (TCIA) have enabled development of new radiomics and machine learning schemes, a key concern remains the generalizability of these methods to unseen datasets. For MRI datasets, model performance could be impacted by (a) site- or scanner-specific variations in image resolution, field-of-view, or image contrast, or (b) presence of imaging artifacts such as noise, motion, inhomogeneity, ringing, or aliasing; which can adversely affect relative image quality between data cohorts. This indicates a need for a quantitative tool to quickly determine relative differences in MRI volumes both within and between large data cohorts. We present MRQy, a new open-source quality control tool to (a) interrogate MRI cohorts for site- or equipment-based differences, and (b) quantify the impact of MRI artifacts on relative image quality; to help determine how to correct for these variations prior to model development. MRQy extracts a series of quality measures (e.g. noise ratios, variation metrics, entropy and energy criteria) and MR image metadata (e.g. voxel resolution, image dimensions) for subsequent interrogation via a specialized HTML5 based front-end designed for real-time filtering and trend visualization. MRQy is designed to be a standalone, unsupervised tool that can be efficiently run on a standard desktop computer. It has been made freely accessible at http://github.com/ccipd/MRQy for wider community use and feedback. MRQy was used to evaluate (a) n=133 brain MRIs from TCIA (7 sites), and (b) n=104 rectal MRIs (3 local sites). MRQy measures revealed significant site-specific variations in both cohorts, indicating potential batch effects. Marked differences in specific MRQy measures were also able to identify MRI datasets that needed to be corrected for common MR imaging artifacts.