 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking community drug response prediction models: datasets, models, tools, and metrics for cross-dataset generalization analysis
Mar 18, 2025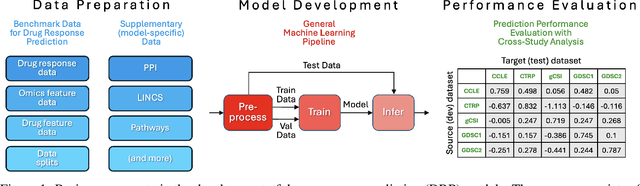
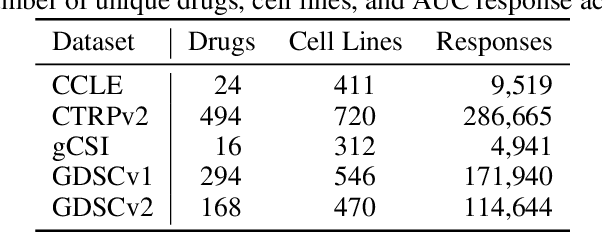
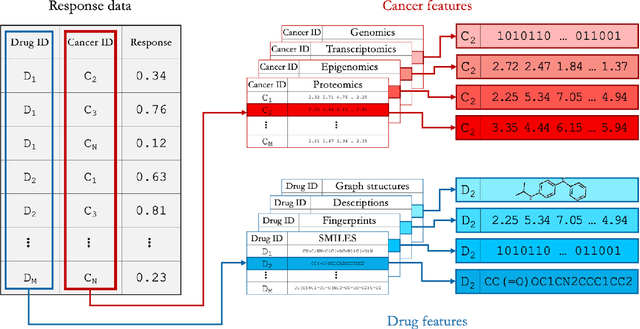

Deep learning (DL) and machine learning (ML) models have shown promise in drug response prediction (DRP), yet their ability to generalize across datasets remains an open question, raising concerns about their real-world applicability. Due to the lack of standardized benchmarking approaches, model evaluations and comparisons often rely on inconsistent datasets and evaluation criteria, making it difficult to assess true predictive capabilities. In this work, we introduce a benchmarking framework for evaluating cross-dataset prediction generalization in DRP models. Our framework incorporates five publicly available drug screening datasets, six standardized DRP models, and a scalable workflow for systematic evaluation. To assess model generalization, we introduce a set of evaluation metrics that quantify both absolute performance (e.g., predictive accuracy across datasets) and relative performance (e.g., performance drop compared to within-dataset results), enabling a more comprehensive assessment of model transferability. Our results reveal substantial performance drops when models are tested on unseen datasets, underscoring the importance of rigorous generalization assessments. While several models demonstrate relatively strong cross-dataset generalization, no single model consistently outperforms across all datasets. Furthermore, we identify CTRPv2 as the most effective source dataset for training, yielding higher generalization scores across target datasets. By sharing this standardized evaluation framework with the community, our study aims to establish a rigorous foundation for model comparison, and accelerate the development of robust DRP models for real-world applications.
Deep learning methods for drug response prediction in cancer: predominant and emerging trends
Nov 18, 2022Cancer claims millions of lives yearly worldwide. While many therapies have been made available in recent years, by in large cancer remains unsolved. Exploiting computational predictive models to study and treat cancer holds great promise in improving drug development and personalized design of treatment plans, ultimately suppressing tumors, alleviating suffering, and prolonging lives of patients. A wave of recent papers demonstrates promising results in predicting cancer response to drug treatments while utilizing deep learning methods. These papers investigate diverse data representations, neural network architectures, learning methodologies, and evaluations schemes. However, deciphering promising predominant and emerging trends is difficult due to the variety of explored methods and lack of standardized framework for comparing drug response prediction models. To obtain a comprehensive landscape of deep learning methods, we conducted an extensive search and analysis of deep learning models that predict the response to single drug treatments. A total of 60 deep learning-based models have been curated and summary plots were generated. Based on the analysis, observable patterns and prevalence of methods have been revealed. This review allows to better understand the current state of the field and identify major challenges and promising solution paths.
Spatial Graph Attention and Curiosity-driven Policy for Antiviral Drug Discovery
Jun 20, 2021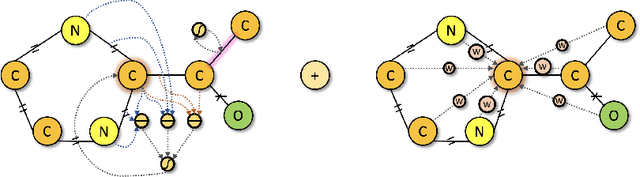
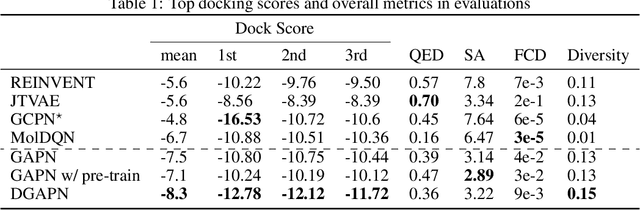
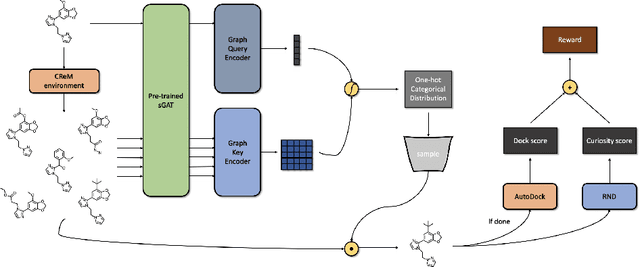
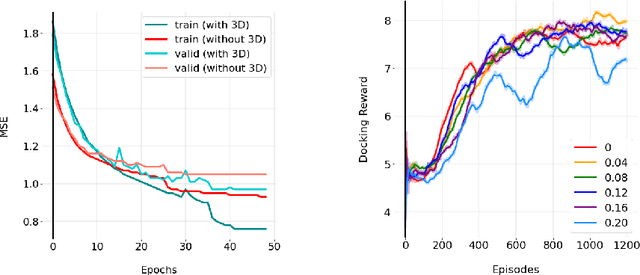
We developed Distilled Graph Attention Policy Networks (DGAPNs), a curiosity-driven reinforcement learning model to generate novel graph-structured chemical representations that optimize user-defined objectives by efficiently navigating a physically constrained domain. The framework is examined on the task of generating molecules that are designed to bind, noncovalently, to functional sites of SARS-CoV-2 proteins. We present a spatial Graph Attention Network (sGAT) that leverages self-attention over both node and edge attributes as well as encoding spatial structure -- this capability is of considerable interest in areas such as molecular and synthetic biology and drug discovery. An attentional policy network is then introduced to learn decision rules for a dynamic, fragment-based chemical environment, and state-of-the-art policy gradient techniques are employed to train the network with enhanced stability. Exploration is efficiently encouraged by incorporating innovation reward bonuses learned and proposed by random network distillation. In experiments, our framework achieved outstanding results compared to state-of-the-art algorithms, while increasing the diversity of proposed molecules and reducing the complexity of paths to chemical synthesis.