 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge14 Examples of How LLMs Can Transform Materials Science and Chemistry: A Reflection on a Large Language Model Hackathon
Jun 13, 2023



Chemistry and materials science are complex. Recently, there have been great successes in addressing this complexity using data-driven or computational techniques. Yet, the necessity of input structured in very specific forms and the fact that there is an ever-growing number of tools creates usability and accessibility challenges. Coupled with the reality that much data in these disciplines is unstructured, the effectiveness of these tools is limited. Motivated by recent works that indicated that large language models (LLMs) might help address some of these issues, we organized a hackathon event on the applications of LLMs in chemistry, materials science, and beyond. This article chronicles the projects built as part of this hackathon. Participants employed LLMs for various applications, including predicting properties of molecules and materials, designing novel interfaces for tools, extracting knowledge from unstructured data, and developing new educational applications. The diverse topics and the fact that working prototypes could be generated in less than two days highlight that LLMs will profoundly impact the future of our fields. The rich collection of ideas and projects also indicates that the applications of LLMs are not limited to materials science and chemistry but offer potential benefits to a wide range of scientific disciplines.
Spatial Graph Attention and Curiosity-driven Policy for Antiviral Drug Discovery
Jun 20, 2021
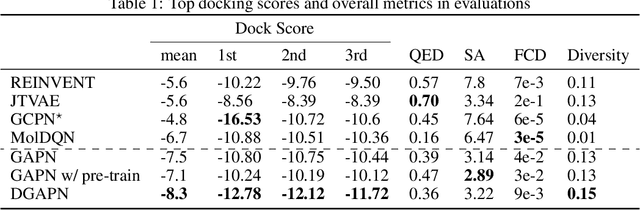
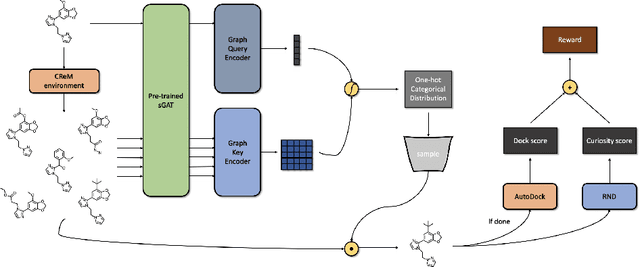
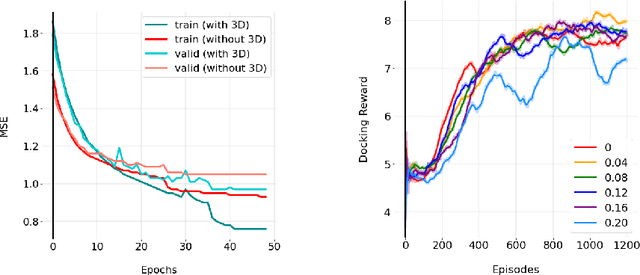
We developed Distilled Graph Attention Policy Networks (DGAPNs), a curiosity-driven reinforcement learning model to generate novel graph-structured chemical representations that optimize user-defined objectives by efficiently navigating a physically constrained domain. The framework is examined on the task of generating molecules that are designed to bind, noncovalently, to functional sites of SARS-CoV-2 proteins. We present a spatial Graph Attention Network (sGAT) that leverages self-attention over both node and edge attributes as well as encoding spatial structure -- this capability is of considerable interest in areas such as molecular and synthetic biology and drug discovery. An attentional policy network is then introduced to learn decision rules for a dynamic, fragment-based chemical environment, and state-of-the-art policy gradient techniques are employed to train the network with enhanced stability. Exploration is efficiently encouraged by incorporating innovation reward bonuses learned and proposed by random network distillation. In experiments, our framework achieved outstanding results compared to state-of-the-art algorithms, while increasing the diversity of proposed molecules and reducing the complexity of paths to chemical synthesis.
Detecting Label Noise via Leave-One-Out Cross-Validation
Mar 28, 2021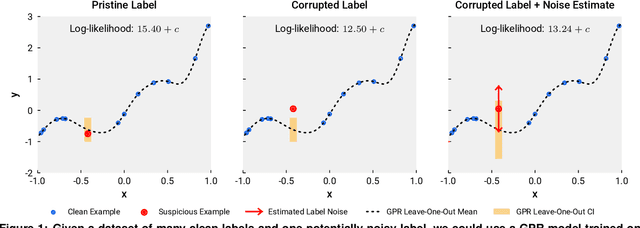
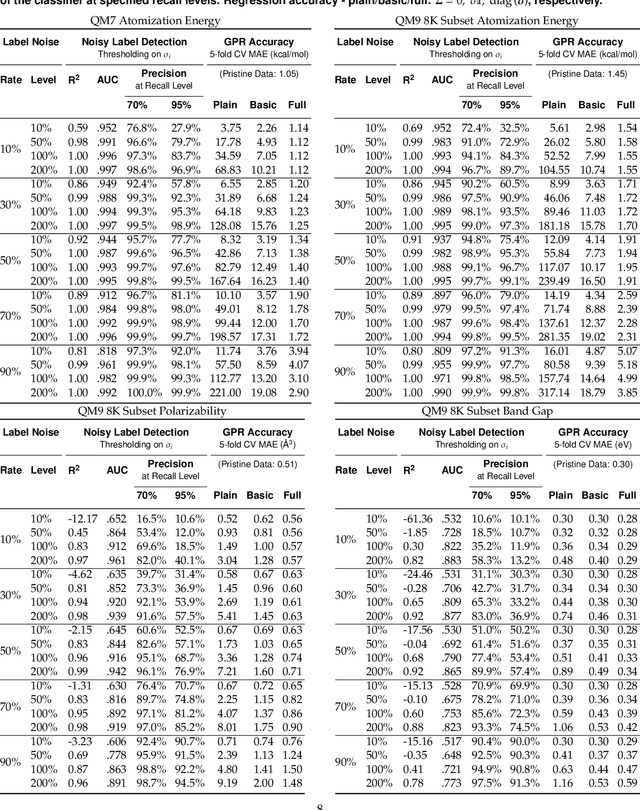
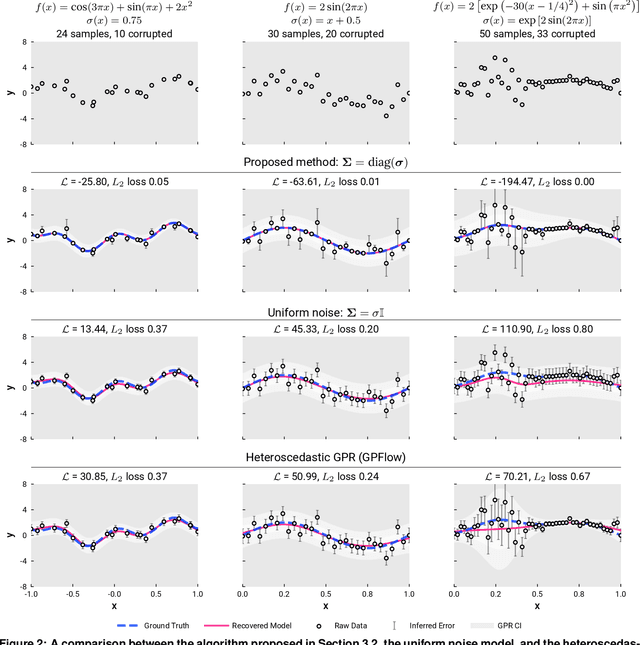
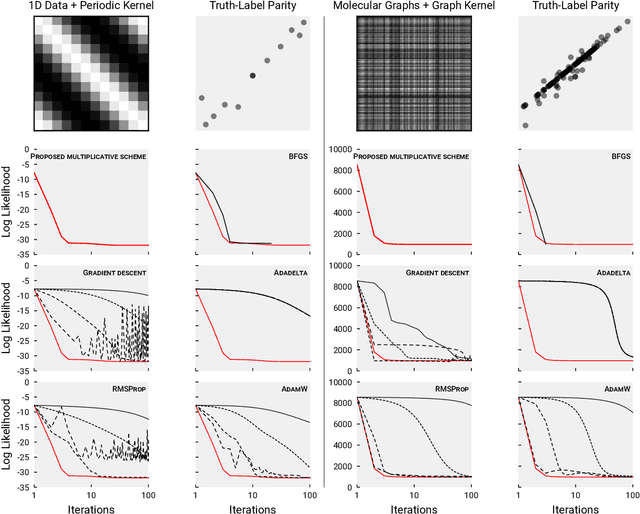
We present a simple algorithm for identifying and correcting real-valued noisy labels from a mixture of clean and corrupted sample points using Gaussian process regression. A heteroscedastic noise model is employed, in which additive Gaussian noise terms with independent variances are associated with each and all of the observed labels. Optimizing the noise model using maximum likelihood estimation leads to the containment of the GPR model's predictive error by the posterior standard deviation in leave-one-out cross-validation. A multiplicative update scheme is proposed for solving the maximum likelihood estimation problem under non-negative constraints. While we provide proof of convergence for certain special cases, the multiplicative scheme has empirically demonstrated monotonic convergence behavior in virtually all our numerical experiments. We show that the presented method can pinpoint corrupted sample points and lead to better regression models when trained on synthetic and real-world scientific data sets.
Prediction of Atomization Energy Using Graph Kernel and Active Learning
Oct 18, 2018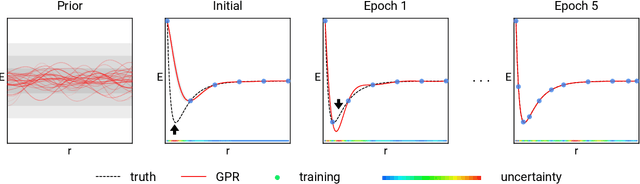
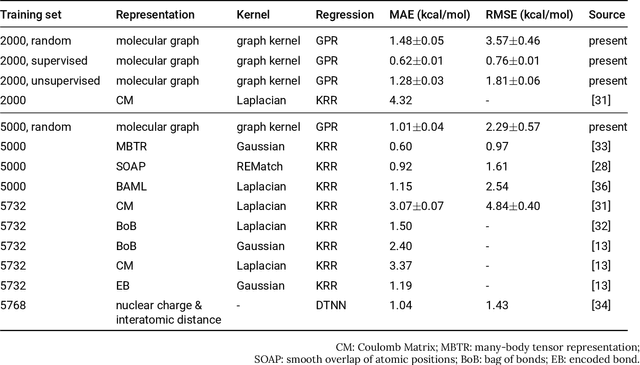
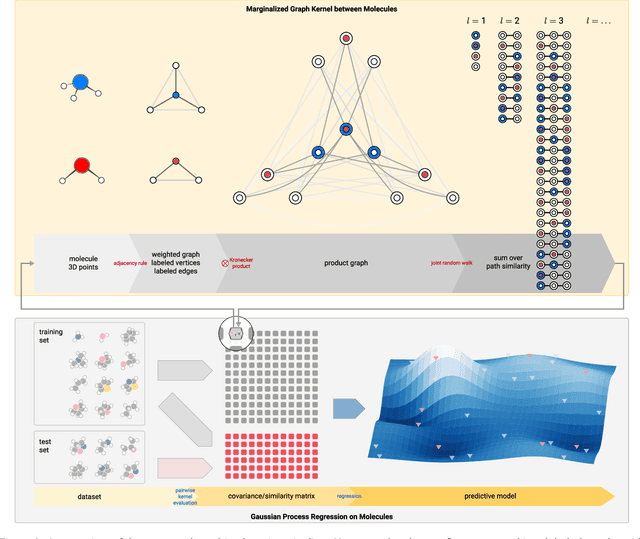
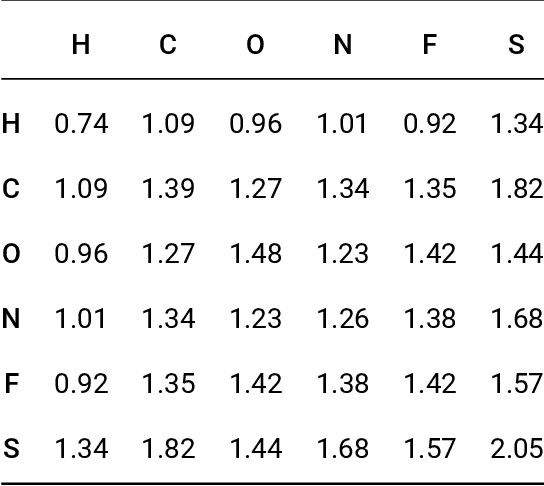
Data-driven prediction of molecular properties presents unique challenges to the design of machine learning methods concerning data structure/dimensionality, symmetry adaption, and confidence management. In this paper, we present a kernel-based pipeline that can learn and predict the atomization energy of molecules with high accuracy. The framework employs Gaussian process regression to perform predictions based on the similarity between molecules, which is computed using the marginalized graph kernel. We discuss why the graph kernel, paired with a graph representation of the molecules, is particularly useful for predicting extensive properties. We demonstrate that using an active learning procedure, the proposed method can achieve a mean absolute error less than 1.0 kcal/mol on the QM7 data set using as few as 1200 training samples and 1 hour of training time. This is a demonstration, in contrast to common believes, that regression models based on kernel methods can be simultaneously accurate and fast predictors.