 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSolving Functional PDEs with Gaussian Processes and Applications to Functional Renormalization Group Equations
Dec 24, 2025We present an operator learning framework for solving non-perturbative functional renormalization group equations, which are integro-differential equations defined on functionals. Our proposed approach uses Gaussian process operator learning to construct a flexible functional representation formulated directly on function space, making it independent of a particular equation or discretization. Our method is flexible, and can apply to a broad range of functional differential equations while still allowing for the incorporation of physical priors in either the prior mean or the kernel design. We demonstrate the performance of our method on several relevant equations, such as the Wetterich and Wilson--Polchinski equations, showing that it achieves equal or better performance than existing approximations such as the local-potential approximation, while being significantly more flexible. In particular, our method can handle non-constant fields, making it promising for the study of more complex field configurations, such as instantons.
Reinforcement Learning Closures for Underresolved Partial Differential Equations using Synthetic Data
May 16, 2025Partial Differential Equations (PDEs) describe phenomena ranging from turbulence and epidemics to quantum mechanics and financial markets. Despite recent advances in computational science, solving such PDEs for real-world applications remains prohibitively expensive because of the necessity of resolving a broad range of spatiotemporal scales. In turn, practitioners often rely on coarse-grained approximations of the original PDEs, trading off accuracy for reduced computational resources. To mitigate the loss of detail inherent in such approximations, closure models are employed to represent unresolved spatiotemporal interactions. We present a framework for developing closure models for PDEs using synthetic data acquired through the method of manufactured solutions. These data are used in conjunction with reinforcement learning to provide closures for coarse-grained PDEs. We illustrate the efficacy of our method using the one-dimensional and two-dimensional Burgers' equations and the two-dimensional advection equation. Moreover, we demonstrate that closure models trained for inhomogeneous PDEs can be effectively generalized to homogeneous PDEs. The results demonstrate the potential for developing accurate and computationally efficient closure models for systems with scarce data.
HiPerRAG: High-Performance Retrieval Augmented Generation for Scientific Insights
May 07, 2025The volume of scientific literature is growing exponentially, leading to underutilized discoveries, duplicated efforts, and limited cross-disciplinary collaboration. Retrieval Augmented Generation (RAG) offers a way to assist scientists by improving the factuality of Large Language Models (LLMs) in processing this influx of information. However, scaling RAG to handle millions of articles introduces significant challenges, including the high computational costs associated with parsing documents and embedding scientific knowledge, as well as the algorithmic complexity of aligning these representations with the nuanced semantics of scientific content. To address these issues, we introduce HiPerRAG, a RAG workflow powered by high performance computing (HPC) to index and retrieve knowledge from more than 3.6 million scientific articles. At its core are Oreo, a high-throughput model for multimodal document parsing, and ColTrast, a query-aware encoder fine-tuning algorithm that enhances retrieval accuracy by using contrastive learning and late-interaction techniques. HiPerRAG delivers robust performance on existing scientific question answering benchmarks and two new benchmarks introduced in this work, achieving 90% accuracy on SciQ and 76% on PubMedQA-outperforming both domain-specific models like PubMedGPT and commercial LLMs such as GPT-4. Scaling to thousands of GPUs on the Polaris, Sunspot, and Frontier supercomputers, HiPerRAG delivers million document-scale RAG workflows for unifying scientific knowledge and fostering interdisciplinary innovation.
Comparative Performance Evaluation of Large Language Models for Extracting Molecular Interactions and Pathway Knowledge
Jul 17, 2023



Understanding protein interactions and pathway knowledge is crucial for unraveling the complexities of living systems and investigating the underlying mechanisms of biological functions and complex diseases. While existing databases provide curated biological data from literature and other sources, they are often incomplete and their maintenance is labor-intensive, necessitating alternative approaches. In this study, we propose to harness the capabilities of large language models to address these issues by automatically extracting such knowledge from the relevant scientific literature. Toward this goal, in this work, we investigate the effectiveness of different large language models in tasks that involve recognizing protein interactions, pathways, and gene regulatory relations. We thoroughly evaluate the performance of various models, highlight the significant findings, and discuss both the future opportunities and the remaining challenges associated with this approach. The code and data are available at: https://github.com/boxorange/BioIE-LLM
Comprehensive analysis of gene expression profiles to radiation exposure reveals molecular signatures of low-dose radiation response
Jan 03, 2023



There are various sources of ionizing radiation exposure, where medical exposure for radiation therapy or diagnosis is the most common human-made source. Understanding how gene expression is modulated after ionizing radiation exposure and investigating the presence of any dose-dependent gene expression patterns have broad implications for health risks from radiotherapy, medical radiation diagnostic procedures, as well as other environmental exposure. In this paper, we perform a comprehensive pathway-based analysis of gene expression profiles in response to low-dose radiation exposure, in order to examine the potential mechanism of gene regulation underlying such responses. To accomplish this goal, we employ a statistical framework to determine whether a specific group of genes belonging to a known pathway display coordinated expression patterns that are modulated in a manner consistent with the radiation level. Findings in our study suggest that there exist complex yet consistent signatures that reflect the molecular response to radiation exposure, which differ between low-dose and high-dose radiation.
Multi-Objective Latent Space Optimization of Generative Molecular Design Models
Mar 01, 2022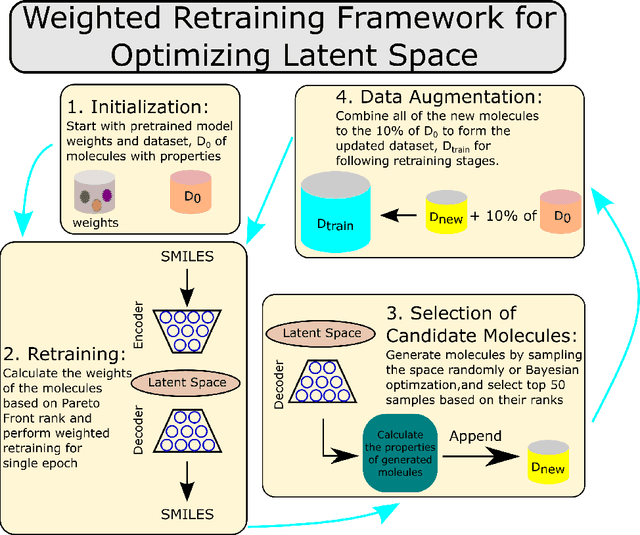
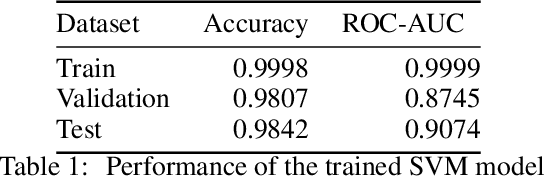
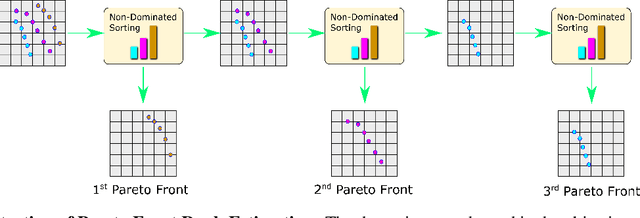
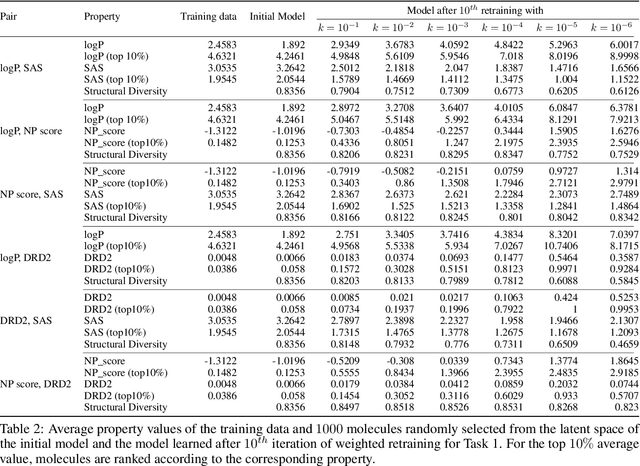
Molecular design based on generative models, such as variational autoencoders (VAEs), has become increasingly popular in recent years due to its efficiency for exploring high-dimensional molecular space to identify molecules with desired properties. While the efficacy of the initial model strongly depends on the training data, the sampling efficiency of the model for suggesting novel molecules with enhanced properties can be further enhanced via latent space optimization. In this paper, we propose a multi-objective latent space optimization (LSO) method that can significantly enhance the performance of generative molecular design (GMD). The proposed method adopts an iterative weighted retraining approach, where the respective weights of the molecules in the training data are determined by their Pareto efficiency. We demonstrate that our multi-objective GMD LSO method can significantly improve the performance of GMD for jointly optimizing multiple molecular properties.
Adaptive Group Testing with Mismatched Models
Oct 05, 2021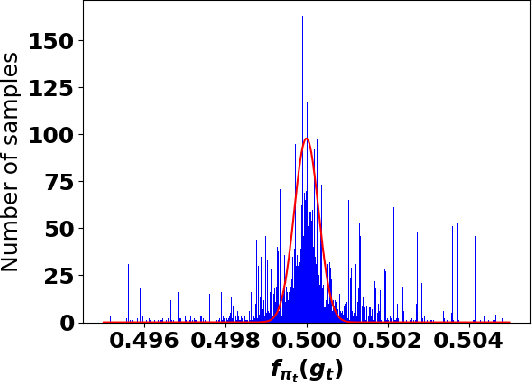
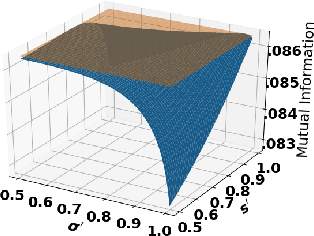
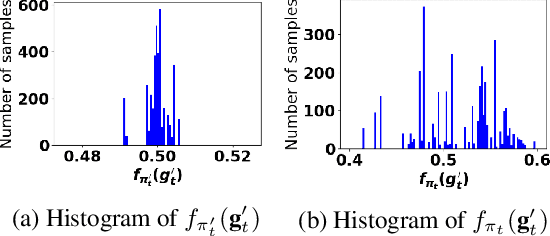
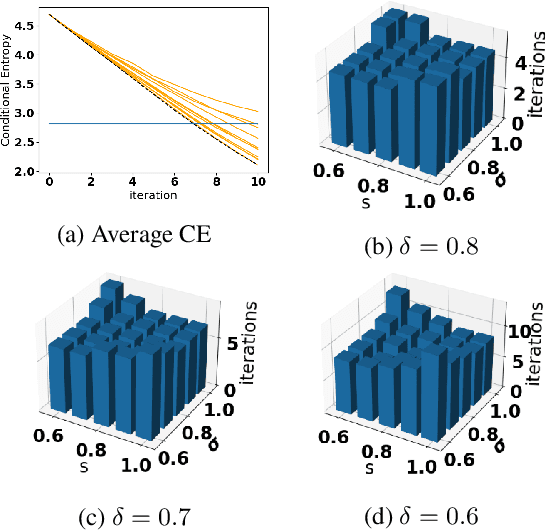
Accurate detection of infected individuals is one of the critical steps in stopping any pandemic. When the underlying infection rate of the disease is low, testing people in groups, instead of testing each individual in the population, can be more efficient. In this work, we consider noisy adaptive group testing design with specific test sensitivity and specificity that select the optimal group given previous test results based on pre-selected utility function. As in prior studies on group testing, we model this problem as a sequential Bayesian Optimal Experimental Design (BOED) to adaptively design the groups for each test. We analyze the required number of group tests when using the updated posterior on the infection status and the corresponding Mutual Information (MI) as our utility function for selecting new groups. More importantly, we study how the potential bias on the ground-truth noise of group tests may affect the group testing sample complexity.
Optimal Decision Making in High-Throughput Virtual Screening Pipelines
Sep 23, 2021
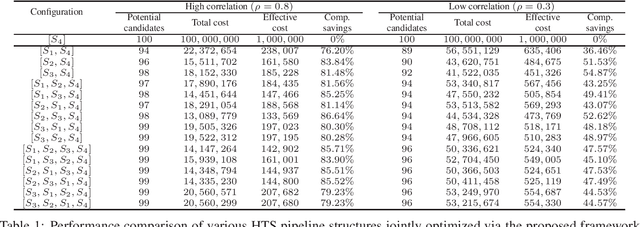
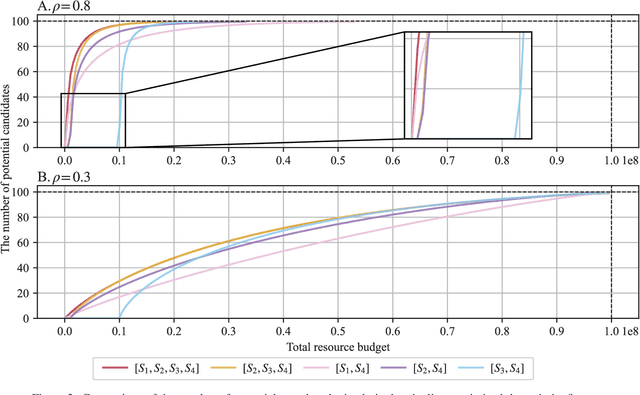
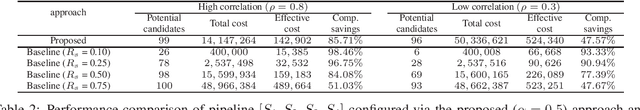
Effective selection of the potential candidates that meet certain conditions in a tremendously large search space has been one of the major concerns in many real-world applications. In addition to the nearly infinitely large search space, rigorous evaluation of a sample based on the reliable experimental or computational platform is often prohibitively expensive, making the screening problem more challenging. In such a case, constructing a high-throughput screening (HTS) pipeline that pre-sifts the samples expected to be potential candidates through the efficient earlier stages, results in a significant amount of savings in resources. However, to the best of our knowledge, despite many successful applications, no one has studied optimal pipeline design or optimal pipeline operations. In this study, we propose two optimization frameworks, applying to most (if not all) screening campaigns involving experimental or/and computational evaluations, for optimally determining the screening thresholds of an HTS pipeline. We validate the proposed frameworks on both analytic and practical scenarios. In particular, we consider the optimal computational campaign for the long non-coding RNA (lncRNA) classification as a practical example. To accomplish this, we built the high-throughput virtual screening (HTVS) pipeline for classifying the lncRNA. The simulation results demonstrate that the proposed frameworks significantly reduce the effective selection cost per potential candidate and make the HTS pipelines less sensitive to their structural variations. In addition to the validation, we provide insights on constructing a better HTS pipeline based on the simulation results.
Robust Importance Sampling for Error Estimation in the Context of Optimal Bayesian Transfer Learning
Sep 05, 2021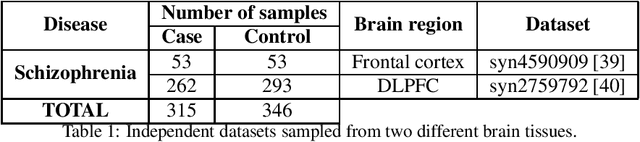
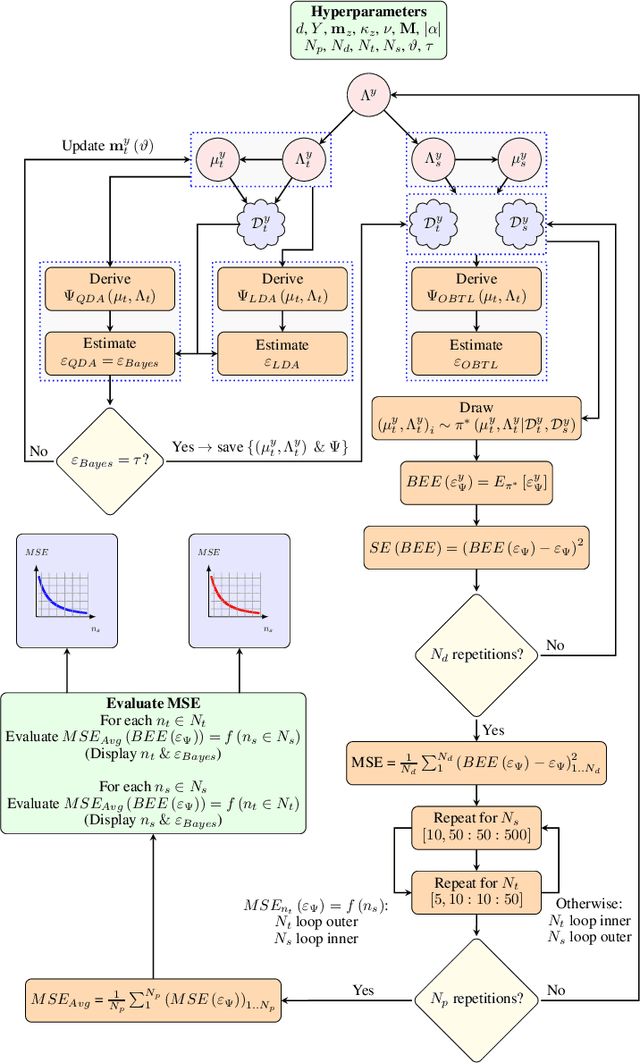
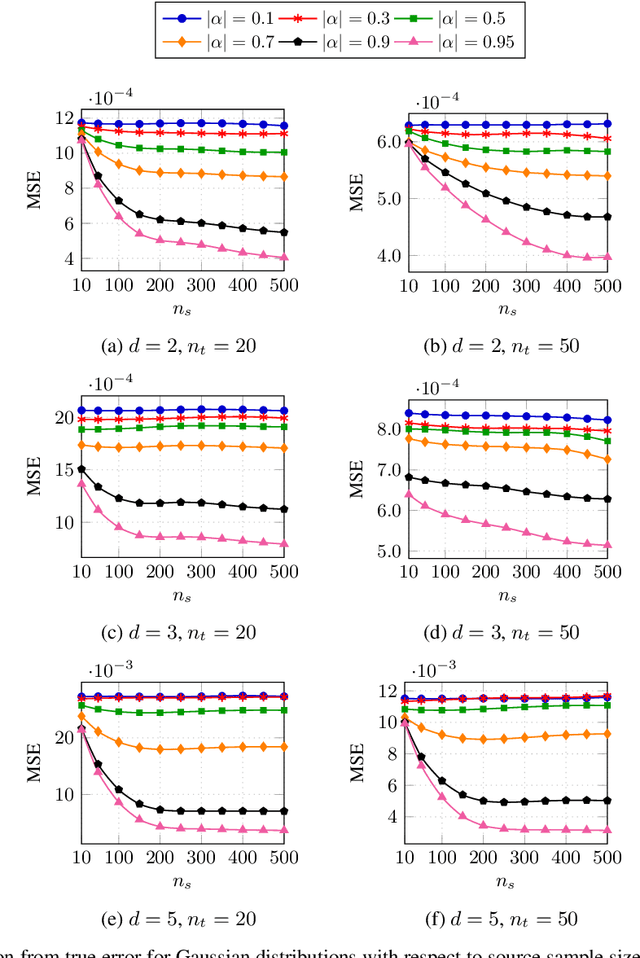
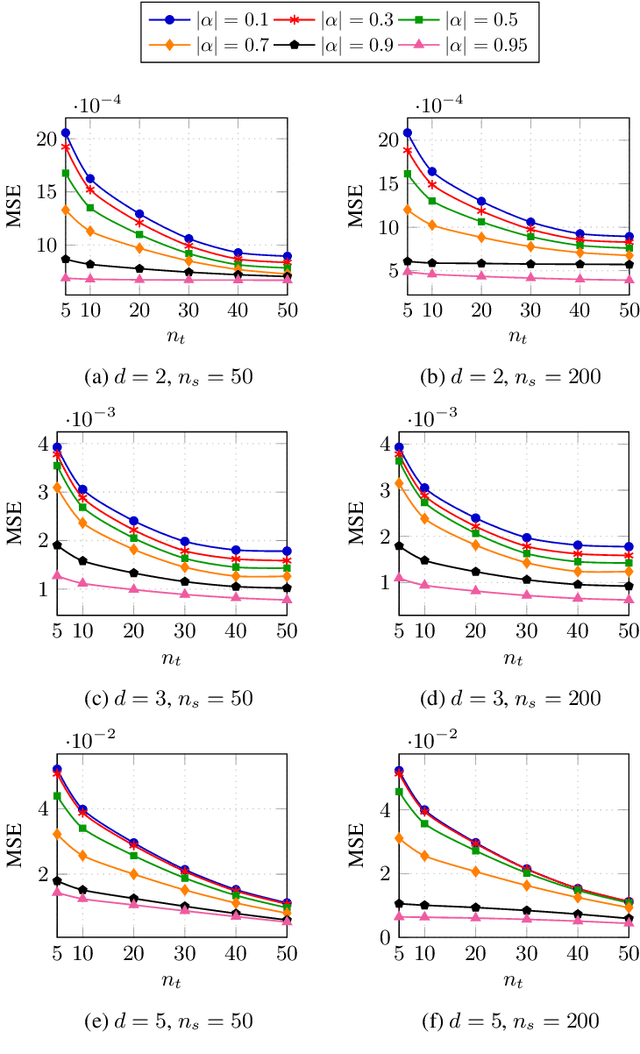
Classification has been a major task for building intelligent systems as it enables decision-making under uncertainty. Classifier design aims at building models from training data for representing feature-label distributions--either explicitly or implicitly. In many scientific or clinical settings, training data are typically limited, which makes designing accurate classifiers and evaluating their classification error extremely challenging. While transfer learning (TL) can alleviate this issue by incorporating data from relevant source domains to improve learning in a different target domain, it has received little attention for performance assessment, notably in error estimation. In this paper, we fill this gap by investigating knowledge transferability in the context of classification error estimation within a Bayesian paradigm. We introduce a novel class of Bayesian minimum mean-square error (MMSE) estimators for optimal Bayesian transfer learning (OBTL), which enables rigorous evaluation of classification error under uncertainty in a small-sample setting. Using Monte Carlo importance sampling, we employ the proposed estimator to evaluate the classification accuracy of a broad family of classifiers that span diverse learning capabilities. Experimental results based on both synthetic data as well as real-world RNA sequencing (RNA-seq) data show that our proposed OBTL error estimation scheme clearly outperforms standard error estimators, especially in a small-sample setting, by tapping into the data from other relevant domains.