 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobust Importance Sampling for Error Estimation in the Context of Optimal Bayesian Transfer Learning
Sep 05, 2021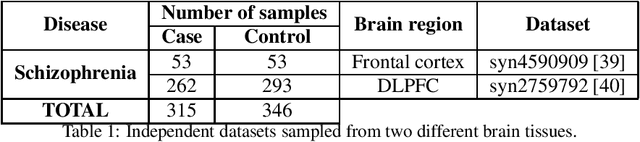
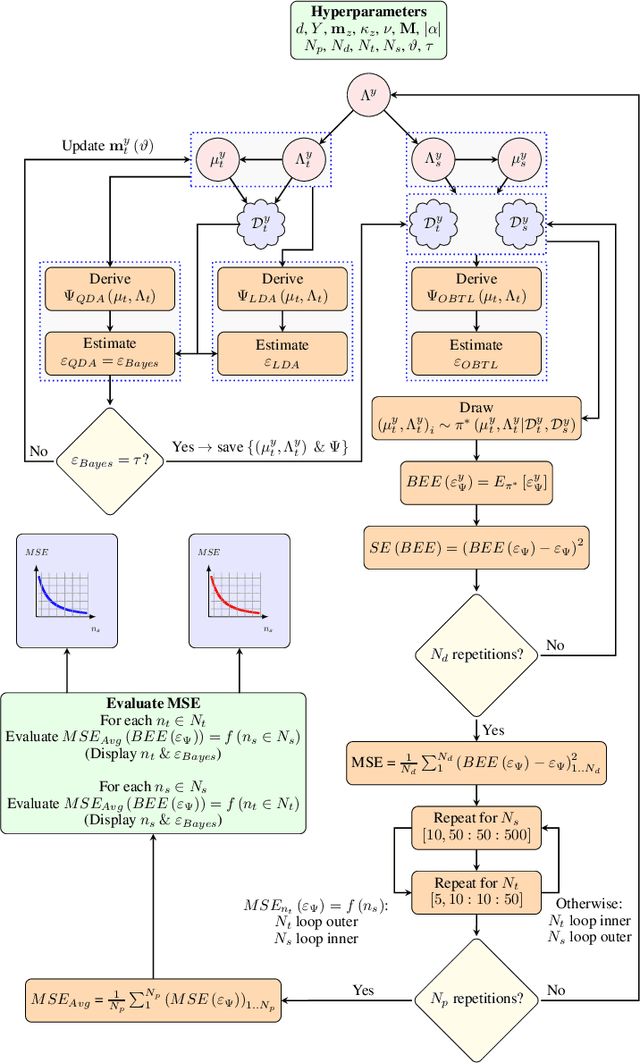
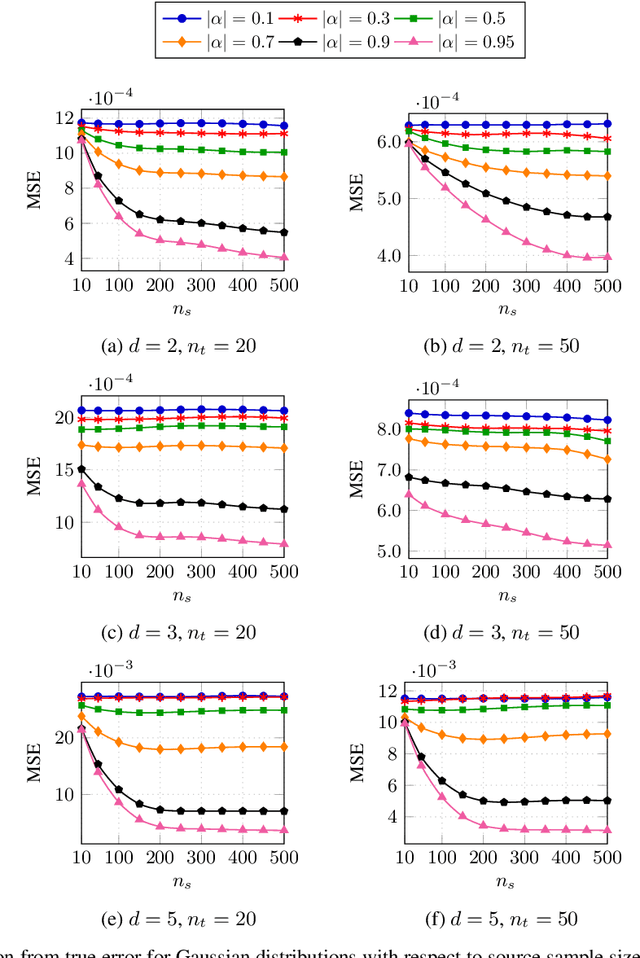
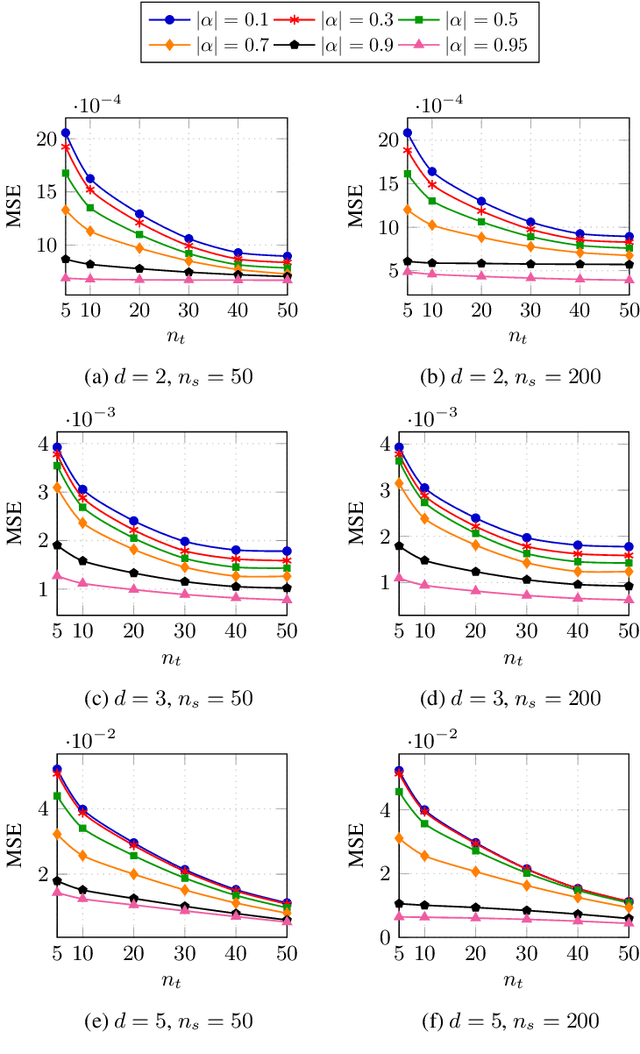
Classification has been a major task for building intelligent systems as it enables decision-making under uncertainty. Classifier design aims at building models from training data for representing feature-label distributions--either explicitly or implicitly. In many scientific or clinical settings, training data are typically limited, which makes designing accurate classifiers and evaluating their classification error extremely challenging. While transfer learning (TL) can alleviate this issue by incorporating data from relevant source domains to improve learning in a different target domain, it has received little attention for performance assessment, notably in error estimation. In this paper, we fill this gap by investigating knowledge transferability in the context of classification error estimation within a Bayesian paradigm. We introduce a novel class of Bayesian minimum mean-square error (MMSE) estimators for optimal Bayesian transfer learning (OBTL), which enables rigorous evaluation of classification error under uncertainty in a small-sample setting. Using Monte Carlo importance sampling, we employ the proposed estimator to evaluate the classification accuracy of a broad family of classifiers that span diverse learning capabilities. Experimental results based on both synthetic data as well as real-world RNA sequencing (RNA-seq) data show that our proposed OBTL error estimation scheme clearly outperforms standard error estimators, especially in a small-sample setting, by tapping into the data from other relevant domains.
Geometric Affinity Propagation for Clustering with Network Knowledge
Mar 26, 2021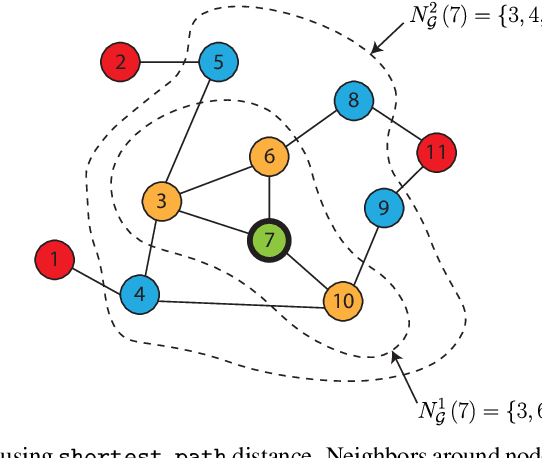
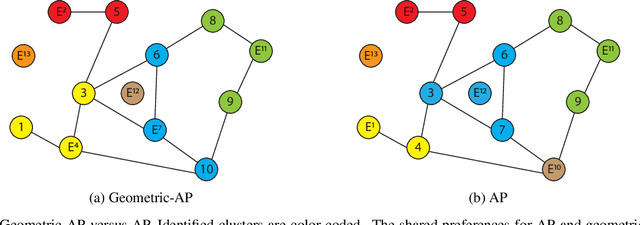
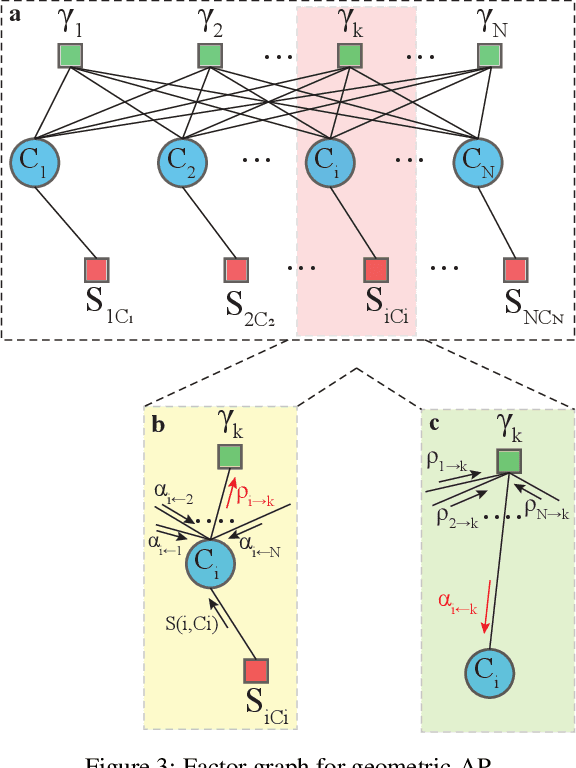
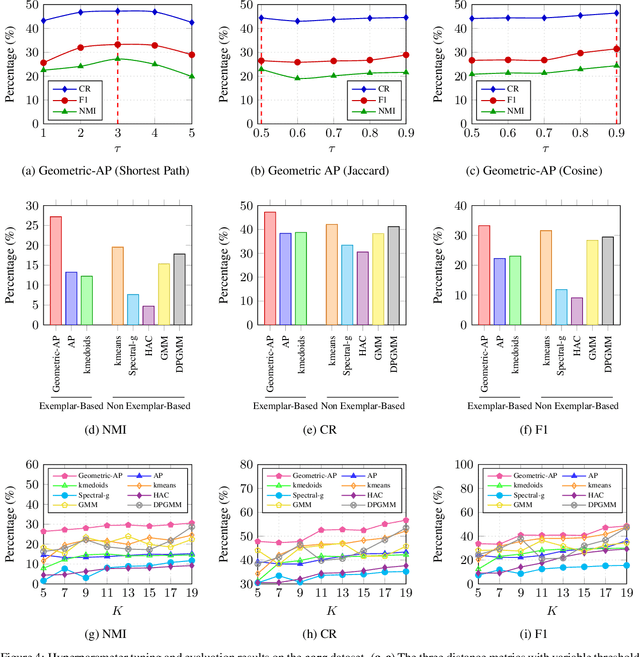
Clustering data into meaningful subsets is a major task in scientific data analysis. To date, various strategies ranging from model-based approaches to data-driven schemes, have been devised for efficient and accurate clustering. One important class of clustering methods that is of a particular interest is the class of exemplar-based approaches. This interest primarily stems from the amount of compressed information encoded in these exemplars that effectively reflect the major characteristics of the respective clusters. Affinity propagation (AP) has proven to be a powerful exemplar-based approach that refines the set of optimal exemplars by iterative pairwise message updates. However, a critical limitation is its inability to capitalize on known networked relations between data points often available for various scientific datasets. To mitigate this shortcoming, we propose geometric-AP, a novel clustering algorithm that effectively extends AP to take advantage of the network topology. Geometric-AP obeys network constraints and uses max-sum belief propagation to leverage the available network topology for generating smooth clusters over the network. Extensive performance assessment reveals a significant enhancement in the quality of the clustering results when compared to benchmark clustering schemes. Especially, we demonstrate that geometric-AP performs extremely well even in cases where the original AP fails drastically.
Neuro-symbolic representation learning on biological knowledge graphs
Dec 13, 2016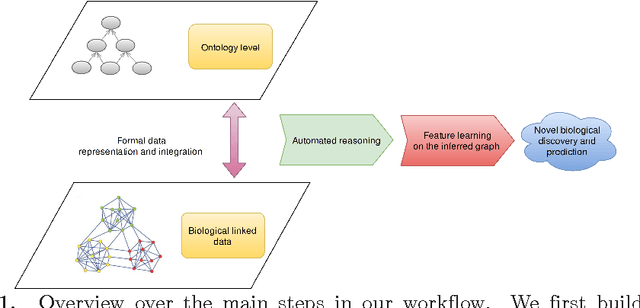
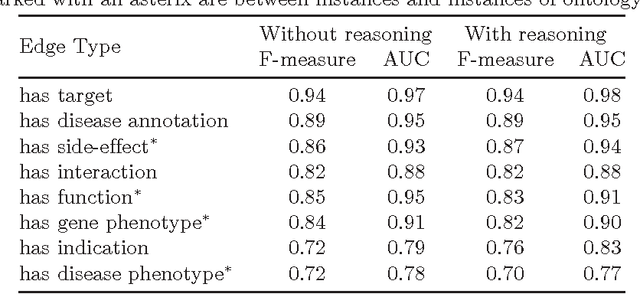
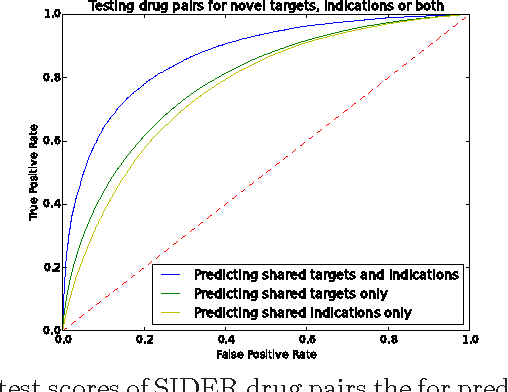
Motivation: Biological data and knowledge bases increasingly rely on Semantic Web technologies and the use of knowledge graphs for data integration, retrieval and federated queries. In the past years, feature learning methods that are applicable to graph-structured data are becoming available, but have not yet widely been applied and evaluated on structured biological knowledge. Results: We develop a novel method for feature learning on biological knowledge graphs. Our method combines symbolic methods, in particular knowledge representation using symbolic logic and automated reasoning, with neural networks to generate embeddings of nodes that encode for related information within knowledge graphs. Through the use of symbolic logic, these embeddings contain both explicit and implicit information. We apply these embeddings to the prediction of edges in the knowledge graph representing problems of function prediction, finding candidate genes of diseases, protein-protein interactions, or drug target relations, and demonstrate performance that matches and sometimes outperforms traditional approaches based on manually crafted features. Our method can be applied to any biological knowledge graph, and will thereby open up the increasing amount of Semantic Web based knowledge bases in biology to use in machine learning and data analytics. Availability and Implementation: https://github.com/bio-ontology-research-group/walking-rdf-and-owl Contact: robert.hoehndorf@kaust.edu.sa