 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeepGO: Predicting protein functions from sequence and interactions using a deep ontology-aware classifier
May 15, 2017

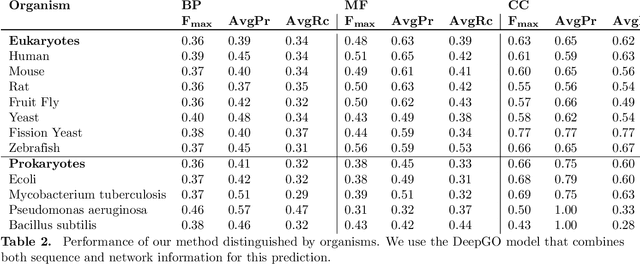

A large number of protein sequences are becoming available through the application of novel high-throughput sequencing technologies. Experimental functional characterization of these proteins is time-consuming and expensive, and is often only done rigorously for few selected model organisms. Computational function prediction approaches have been suggested to fill this gap. The functions of proteins are classified using the Gene Ontology (GO), which contains over 40,000 classes. Additionally, proteins have multiple functions, making function prediction a large-scale, multi-class, multi-label problem. We have developed a novel method to predict protein function from sequence. We use deep learning to learn features from protein sequences as well as a cross-species protein-protein interaction network. Our approach specifically outputs information in the structure of the GO and utilizes the dependencies between GO classes as background information to construct a deep learning model. We evaluate our method using the standards established by the Computational Assessment of Function Annotation (CAFA) and demonstrate a significant improvement over baseline methods such as BLAST, with significant improvement for predicting cellular locations.
Neuro-symbolic representation learning on biological knowledge graphs
Dec 13, 2016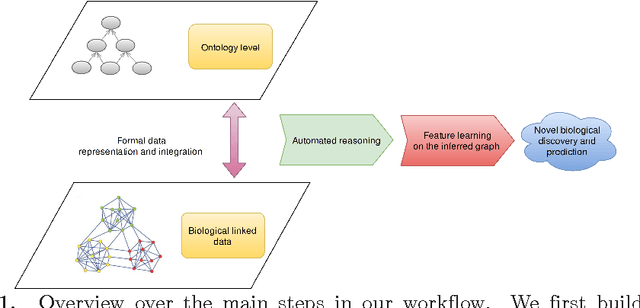
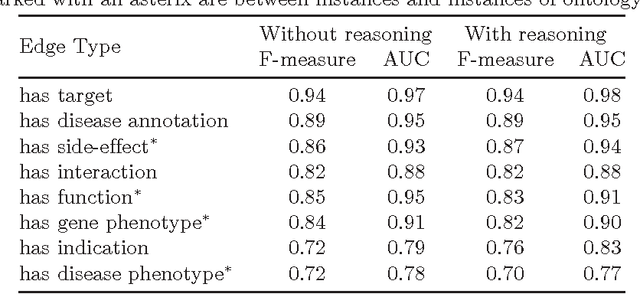
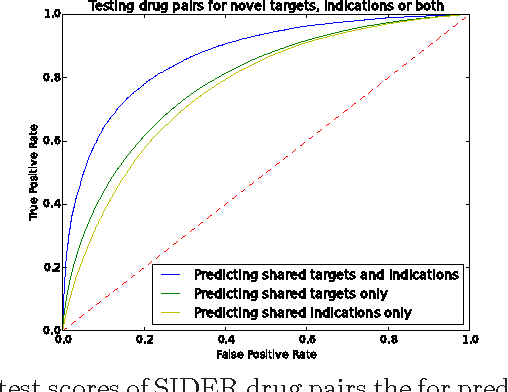
Motivation: Biological data and knowledge bases increasingly rely on Semantic Web technologies and the use of knowledge graphs for data integration, retrieval and federated queries. In the past years, feature learning methods that are applicable to graph-structured data are becoming available, but have not yet widely been applied and evaluated on structured biological knowledge. Results: We develop a novel method for feature learning on biological knowledge graphs. Our method combines symbolic methods, in particular knowledge representation using symbolic logic and automated reasoning, with neural networks to generate embeddings of nodes that encode for related information within knowledge graphs. Through the use of symbolic logic, these embeddings contain both explicit and implicit information. We apply these embeddings to the prediction of edges in the knowledge graph representing problems of function prediction, finding candidate genes of diseases, protein-protein interactions, or drug target relations, and demonstrate performance that matches and sometimes outperforms traditional approaches based on manually crafted features. Our method can be applied to any biological knowledge graph, and will thereby open up the increasing amount of Semantic Web based knowledge bases in biology to use in machine learning and data analytics. Availability and Implementation: https://github.com/bio-ontology-research-group/walking-rdf-and-owl Contact: robert.hoehndorf@kaust.edu.sa