 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFinal Report for CHESS: Cloud, High-Performance Computing, and Edge for Science and Security
Oct 21, 2024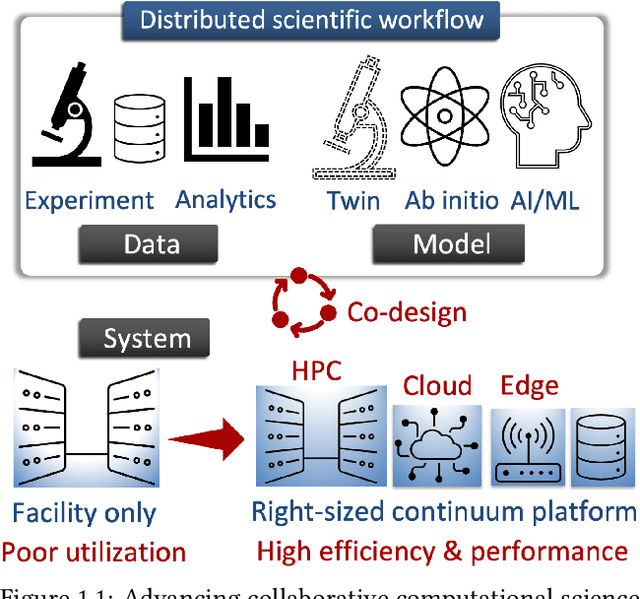
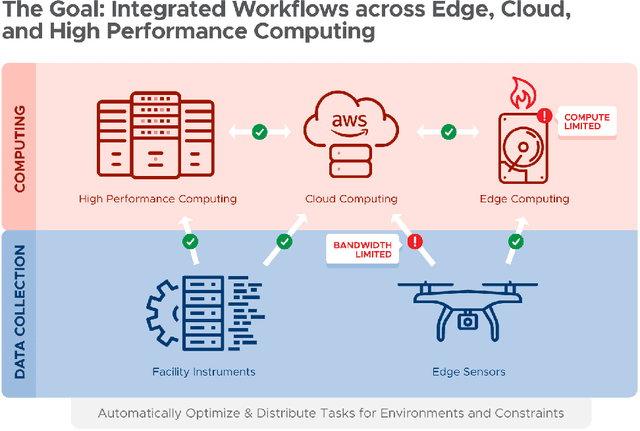
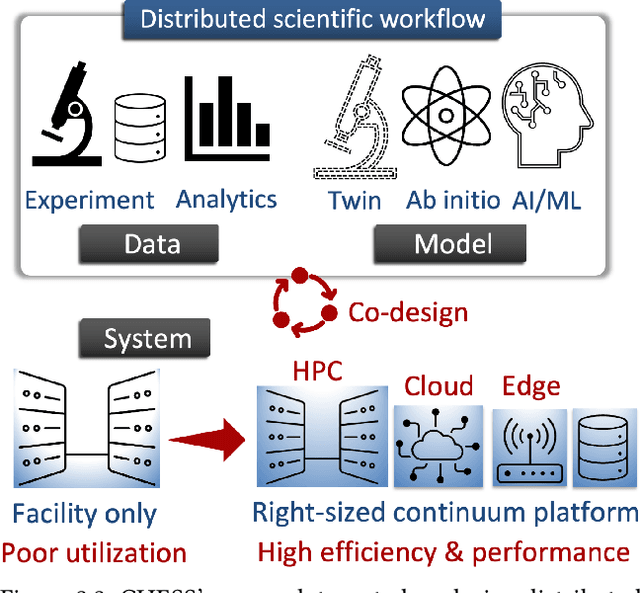

Automating the theory-experiment cycle requires effective distributed workflows that utilize a computing continuum spanning lab instruments, edge sensors, computing resources at multiple facilities, data sets distributed across multiple information sources, and potentially cloud. Unfortunately, the obvious methods for constructing continuum platforms, orchestrating workflow tasks, and curating datasets over time fail to achieve scientific requirements for performance, energy, security, and reliability. Furthermore, achieving the best use of continuum resources depends upon the efficient composition and execution of workflow tasks, i.e., combinations of numerical solvers, data analytics, and machine learning. Pacific Northwest National Laboratory's LDRD "Cloud, High-Performance Computing (HPC), and Edge for Science and Security" (CHESS) has developed a set of interrelated capabilities for enabling distributed scientific workflows and curating datasets. This report describes the results and successes of CHESS from the perspective of open science.
Achieving 100X faster simulations of complex biological phenomena by coupling ML to HPC ensembles
Apr 26, 2021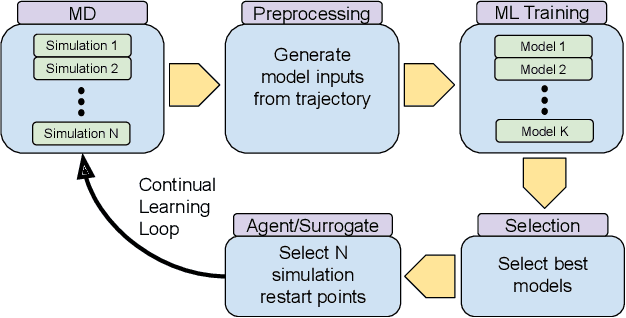
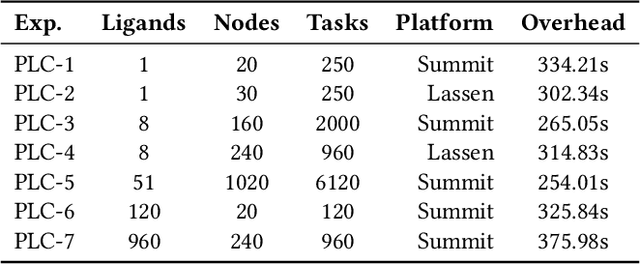
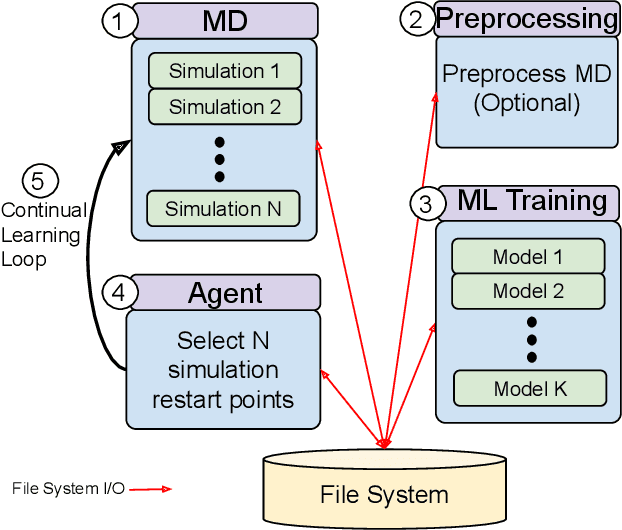
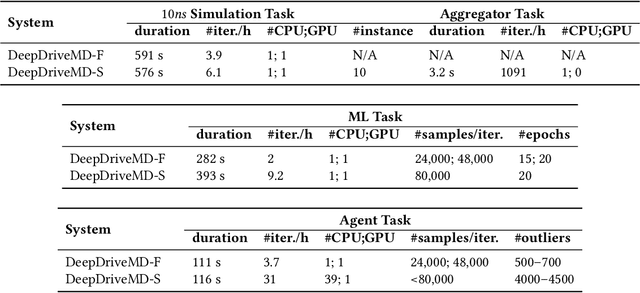
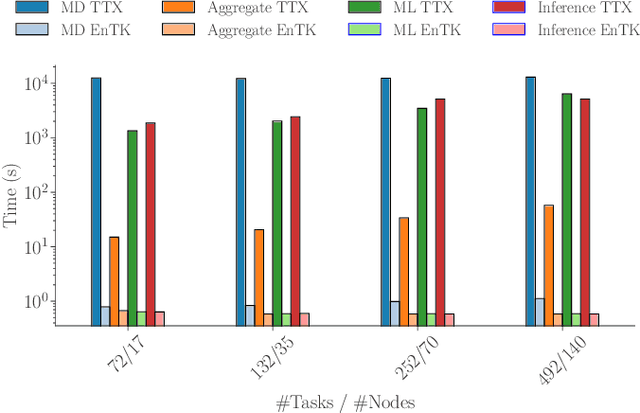
The use of ML methods to dynamically steer ensemble-based simulations promises significant improvements in the performance of scientific applications. We present DeepDriveMD, a tool for a range of prototypical ML-driven HPC simulation scenarios, and use it to quantify improvements in the scientific performance of ML-driven ensemble-based applications. We discuss its design and characterize its performance. Motivated by the potential for further scientific improvements and applicability to more sophisticated physical systems, we extend the design of DeepDriveMD to support stream-based communication between simulations and learning methods. It demonstrates a 100x speedup to fold proteins, and performs 1.6x more simulations per unit time, improving resource utilization compared to the sequential framework. Experiments are performed on leadership-class platforms, at scales of up to O(1000) nodes, and for production workloads. We establish DeepDriveMD as a high-performance framework for ML-driven HPC simulation scenarios, that supports diverse simulation and ML back-ends, and which enables new scientific insights by improving length- and time-scale accessed.
DeepDriveMD: Deep-Learning Driven Adaptive Molecular Simulations for Protein Folding
Sep 17, 2019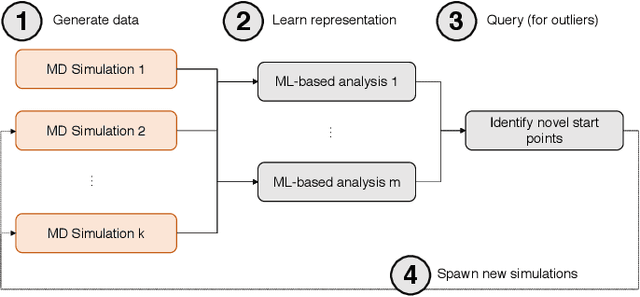
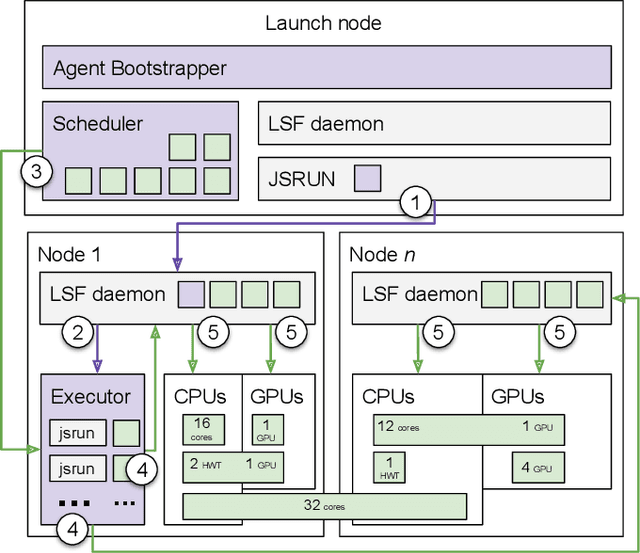
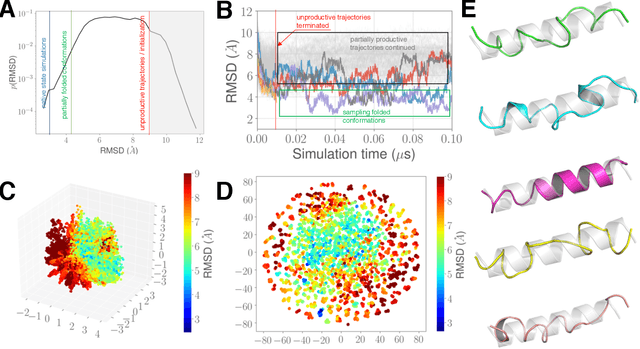
Simulations of biological macromolecules play an important role in understanding the physical basis of a number of complex processes such as protein folding. Even with increasing computational power and evolution of specialized architectures, the ability to simulate protein folding at atomistic scales still remains challenging. This stems from the dual aspects of high dimensionality of protein conformational landscapes, and the inability of atomistic molecular dynamics (MD) simulations to sufficiently sample these landscapes to observe folding events. Machine learning/deep learning (ML/DL) techniques, when combined with atomistic MD simulations offer the opportunity to potentially overcome these limitations by: (1) effectively reducing the dimensionality of MD simulations to automatically build latent representations that correspond to biophysically relevant reaction coordinates (RCs), and (2) driving MD simulations to automatically sample potentially novel conformational states based on these RCs. We examine how coupling DL approaches with MD simulations can fold small proteins effectively on supercomputers. In particular, we study the computational costs and effectiveness of scaling DL-coupled MD workflows by folding two prototypical systems, viz., Fs-peptide and the fast-folding variant of the villin head piece protein. We demonstrate that a DL driven MD workflow is able to effectively learn latent representations and drive adaptive simulations. Compared to traditional MD-based approaches, our approach achieves an effective performance gain in sampling the folded states by at least 2.3x. Our study provides a quantitative basis to understand how DL driven MD simulations, can lead to effective performance gains and reduced times to solution on supercomputing resources.