 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTransferring a molecular foundation model for polymer property predictions
Oct 25, 2023Transformer-based large language models have remarkable potential to accelerate design optimization for applications such as drug development and materials discovery. Self-supervised pretraining of transformer models requires large-scale datasets, which are often sparsely populated in topical areas such as polymer science. State-of-the-art approaches for polymers conduct data augmentation to generate additional samples but unavoidably incurs extra computational costs. In contrast, large-scale open-source datasets are available for small molecules and provide a potential solution to data scarcity through transfer learning. In this work, we show that using transformers pretrained on small molecules and fine-tuned on polymer properties achieve comparable accuracy to those trained on augmented polymer datasets for a series of benchmark prediction tasks.
ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High Performance Computing
Jul 20, 2020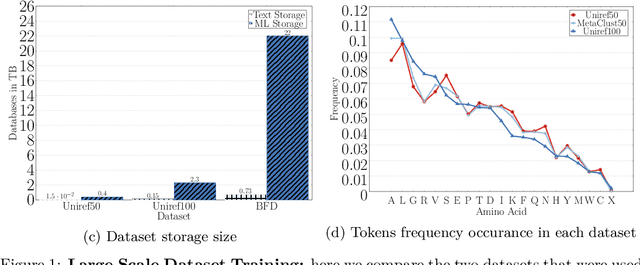
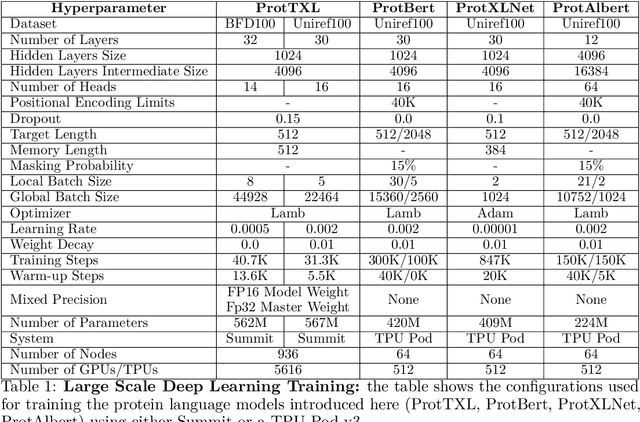
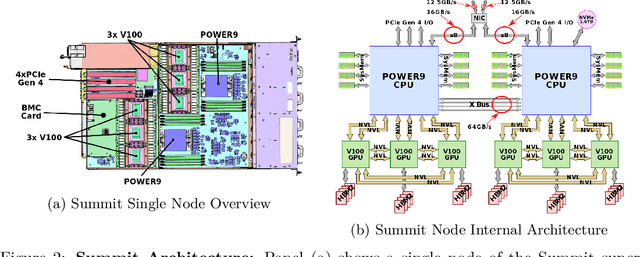
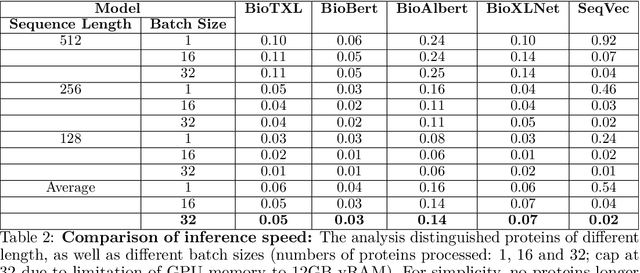
Computational biology and bioinformatics provide vast data gold-mines from protein sequences, ideal for Language Models (LMs) taken from Natural Language Processing (NLP). These LMs reach for new prediction frontiers at low inference costs. Here, we trained two auto-regressive language models (Transformer-XL, XLNet) and two auto-encoder models (Bert, Albert) on data from UniRef and BFD containing up to 393 billion amino acids (words) from 2.1 billion protein sequences (22- and 112-times the entire English Wikipedia). The LMs were trained on the Summit supercomputer at Oak Ridge National Laboratory (ORNL), using 936 nodes (total 5616 GPUs) and one TPU Pod (V3-512 or V3-1024). We validated the advantage of up-scaling LMs to larger models supported by bigger data by predicting secondary structure (3-states: Q3=76-84, 8-states: Q8=65-73), sub-cellular localization for 10 cellular compartments (Q10=74) and whether a protein is membrane-bound or water-soluble (Q2=89). Dimensionality reduction revealed that the LM-embeddings from unlabeled data (only protein sequences) captured important biophysical properties governing protein shape. This implied learning some of the grammar of the language of life realized in protein sequences. The successful up-scaling of protein LMs through HPC to larger data sets slightly reduced the gap between models trained on evolutionary information and LMs. The official GitHub repository: https://github.com/agemagician/ProtTrans
DeepDriveMD: Deep-Learning Driven Adaptive Molecular Simulations for Protein Folding
Sep 17, 2019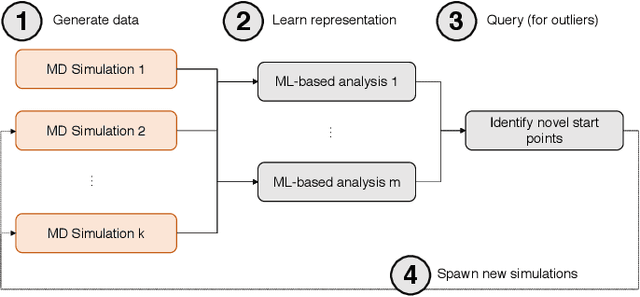
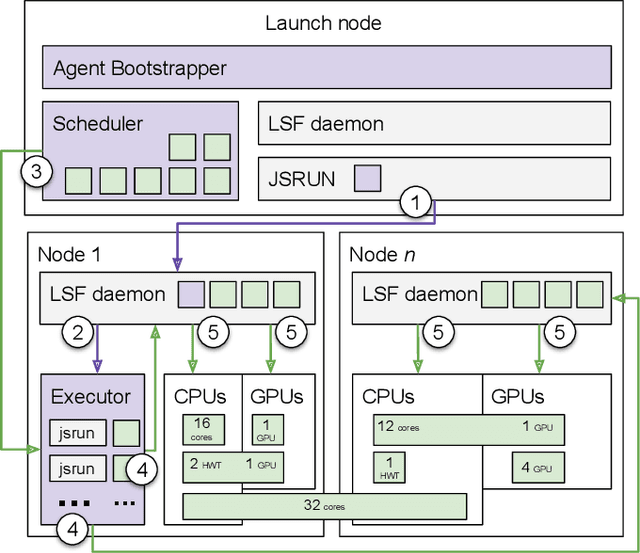
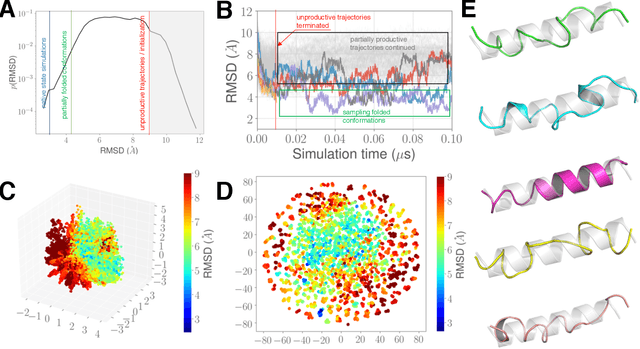
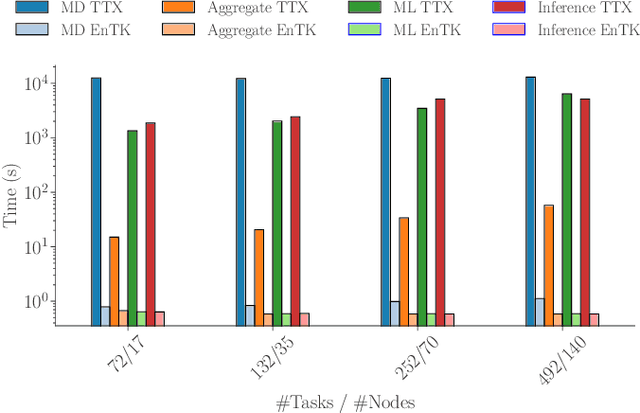
Simulations of biological macromolecules play an important role in understanding the physical basis of a number of complex processes such as protein folding. Even with increasing computational power and evolution of specialized architectures, the ability to simulate protein folding at atomistic scales still remains challenging. This stems from the dual aspects of high dimensionality of protein conformational landscapes, and the inability of atomistic molecular dynamics (MD) simulations to sufficiently sample these landscapes to observe folding events. Machine learning/deep learning (ML/DL) techniques, when combined with atomistic MD simulations offer the opportunity to potentially overcome these limitations by: (1) effectively reducing the dimensionality of MD simulations to automatically build latent representations that correspond to biophysically relevant reaction coordinates (RCs), and (2) driving MD simulations to automatically sample potentially novel conformational states based on these RCs. We examine how coupling DL approaches with MD simulations can fold small proteins effectively on supercomputers. In particular, we study the computational costs and effectiveness of scaling DL-coupled MD workflows by folding two prototypical systems, viz., Fs-peptide and the fast-folding variant of the villin head piece protein. We demonstrate that a DL driven MD workflow is able to effectively learn latent representations and drive adaptive simulations. Compared to traditional MD-based approaches, our approach achieves an effective performance gain in sampling the folded states by at least 2.3x. Our study provides a quantitative basis to understand how DL driven MD simulations, can lead to effective performance gains and reduced times to solution on supercomputing resources.