 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaling Atomistic Protein Binder Design with Generative Pretraining and Test-Time Compute
Mar 30, 2026Protein interaction modeling is central to protein design, which has been transformed by machine learning with applications in drug discovery and beyond. In this landscape, structure-based de novo binder design is cast as either conditional generative modeling or sequence optimization via structure predictors ("hallucination"). We argue that this is a false dichotomy and propose Proteina-Complexa, a novel fully atomistic binder generation method unifying both paradigms. We extend recent flow-based latent protein generation architectures and leverage the domain-domain interactions of monomeric computationally predicted protein structures to construct Teddymer, a new large-scale dataset of synthetic binder-target pairs for pretraining. Combined with high-quality experimental multimers, this enables training a strong base model. We then perform inference-time optimization with this generative prior, unifying the strengths of previously distinct generative and hallucination methods. Proteina-Complexa sets a new state of the art in computational binder design benchmarks: it delivers markedly higher in-silico success rates than existing generative approaches, and our novel test-time optimization strategies greatly outperform previous hallucination methods under normalized compute budgets. We also demonstrate interface hydrogen bond optimization, fold class-guided binder generation, and extensions to small molecule targets and enzyme design tasks, again surpassing prior methods. Code, models and new data will be publicly released.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024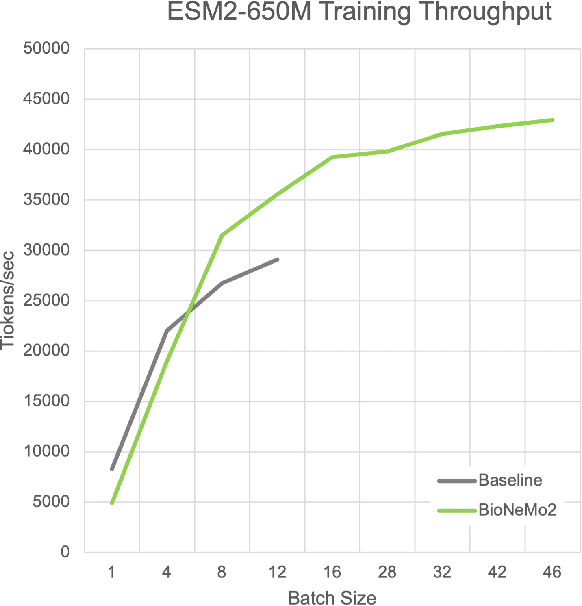
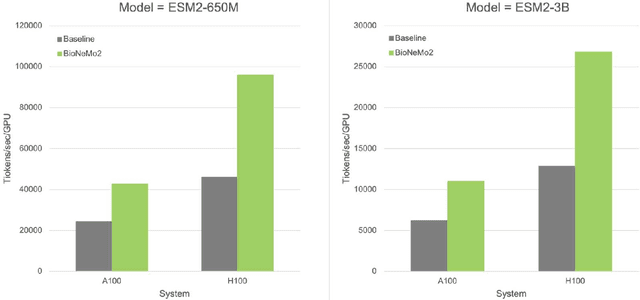
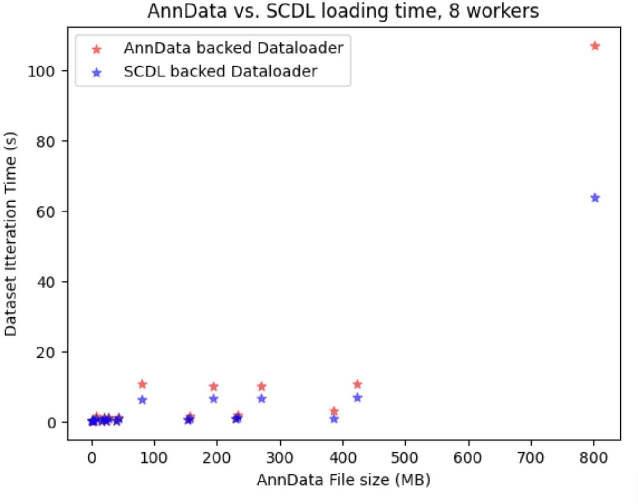
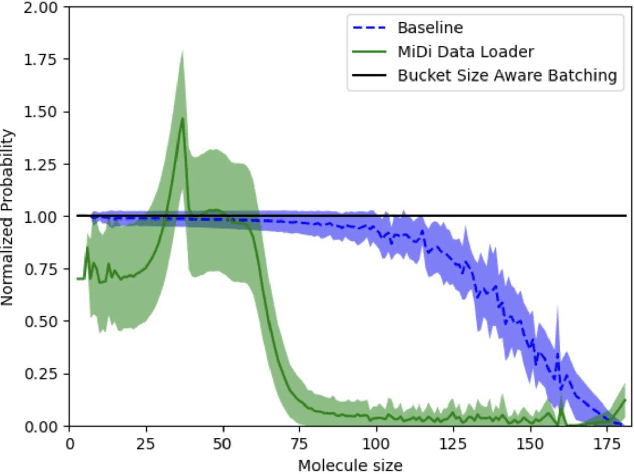
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High Performance Computing
Jul 20, 2020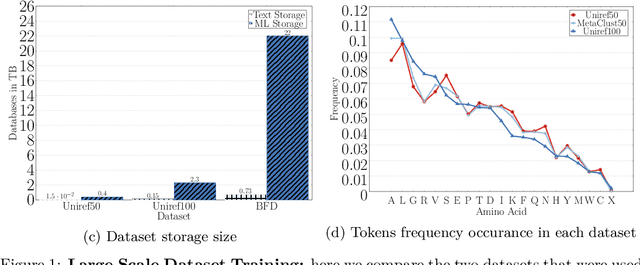
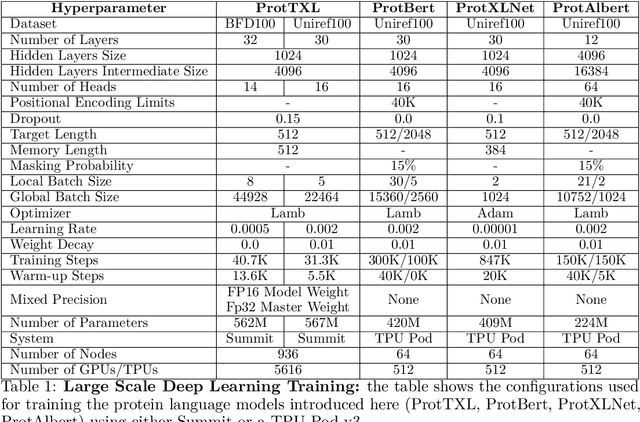
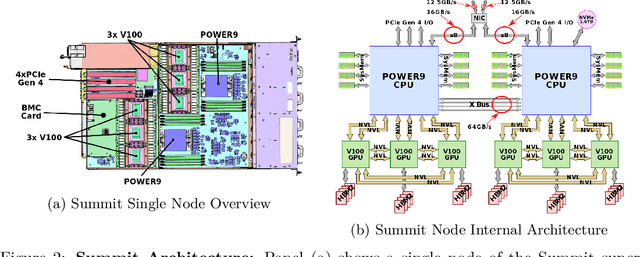
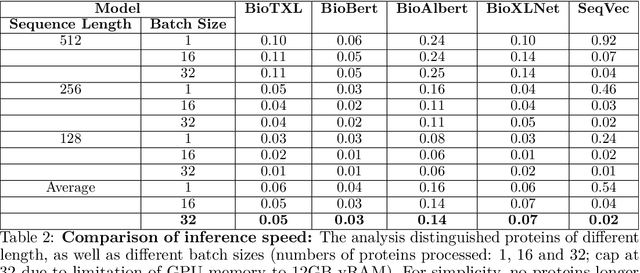
Computational biology and bioinformatics provide vast data gold-mines from protein sequences, ideal for Language Models (LMs) taken from Natural Language Processing (NLP). These LMs reach for new prediction frontiers at low inference costs. Here, we trained two auto-regressive language models (Transformer-XL, XLNet) and two auto-encoder models (Bert, Albert) on data from UniRef and BFD containing up to 393 billion amino acids (words) from 2.1 billion protein sequences (22- and 112-times the entire English Wikipedia). The LMs were trained on the Summit supercomputer at Oak Ridge National Laboratory (ORNL), using 936 nodes (total 5616 GPUs) and one TPU Pod (V3-512 or V3-1024). We validated the advantage of up-scaling LMs to larger models supported by bigger data by predicting secondary structure (3-states: Q3=76-84, 8-states: Q8=65-73), sub-cellular localization for 10 cellular compartments (Q10=74) and whether a protein is membrane-bound or water-soluble (Q2=89). Dimensionality reduction revealed that the LM-embeddings from unlabeled data (only protein sequences) captured important biophysical properties governing protein shape. This implied learning some of the grammar of the language of life realized in protein sequences. The successful up-scaling of protein LMs through HPC to larger data sets slightly reduced the gap between models trained on evolutionary information and LMs. The official GitHub repository: https://github.com/agemagician/ProtTrans