 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024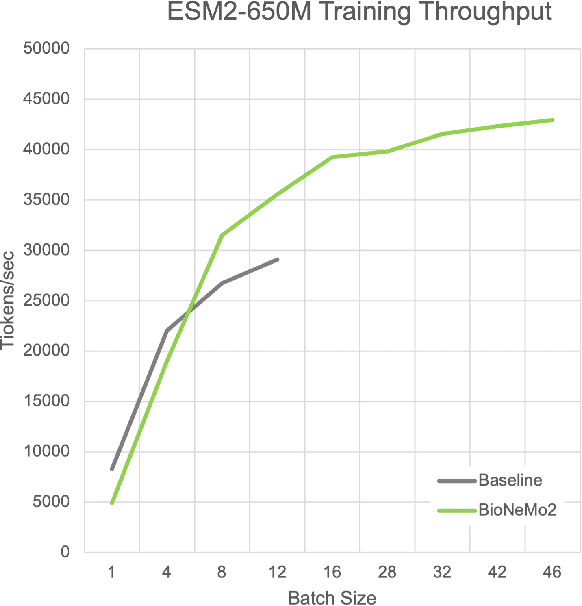
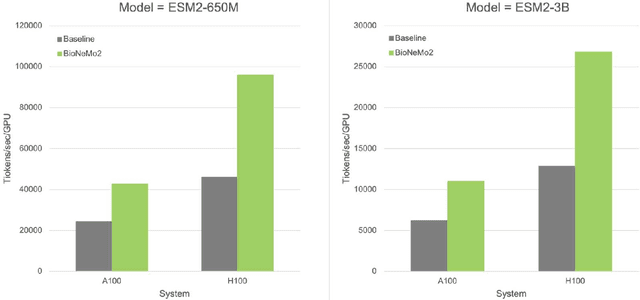
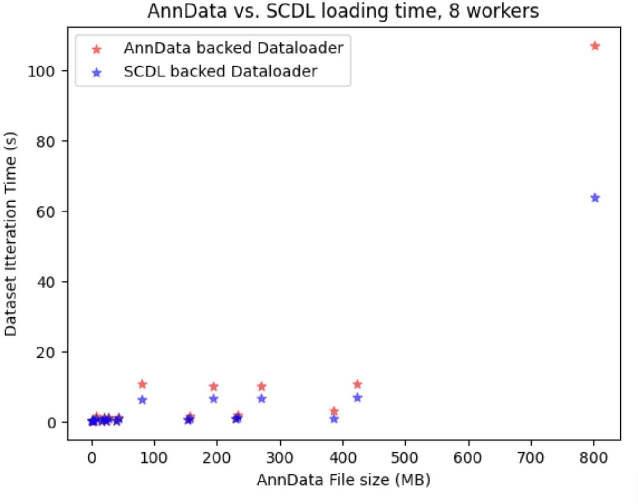
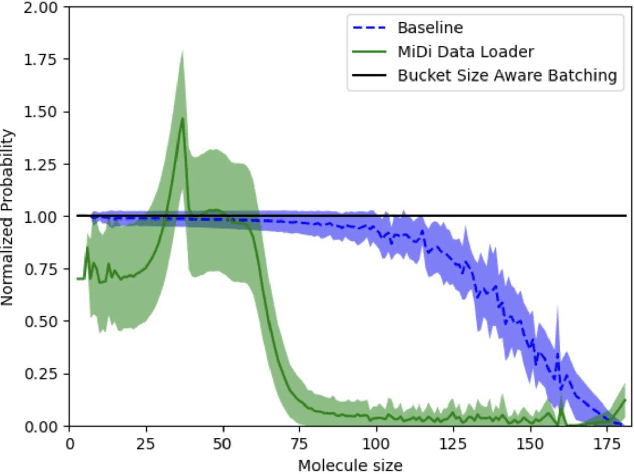
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.
DeepSpeed4Science Initiative: Enabling Large-Scale Scientific Discovery through Sophisticated AI System Technologies
Oct 11, 2023



In the upcoming decade, deep learning may revolutionize the natural sciences, enhancing our capacity to model and predict natural occurrences. This could herald a new era of scientific exploration, bringing significant advancements across sectors from drug development to renewable energy. To answer this call, we present DeepSpeed4Science initiative (deepspeed4science.ai) which aims to build unique capabilities through AI system technology innovations to help domain experts to unlock today's biggest science mysteries. By leveraging DeepSpeed's current technology pillars (training, inference and compression) as base technology enablers, DeepSpeed4Science will create a new set of AI system technologies tailored for accelerating scientific discoveries by addressing their unique complexity beyond the common technical approaches used for accelerating generic large language models (LLMs). In this paper, we showcase the early progress we made with DeepSpeed4Science in addressing two of the critical system challenges in structural biology research.
3D Infomax improves GNNs for Molecular Property Prediction
Oct 08, 2021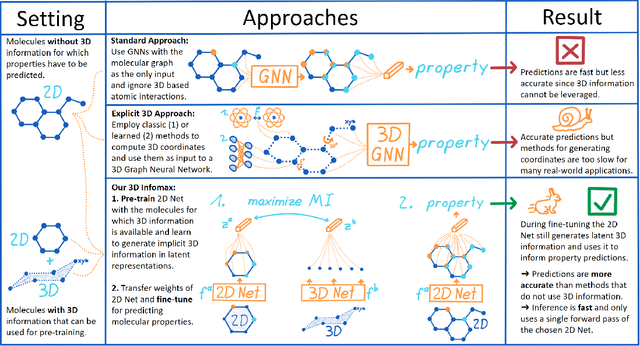
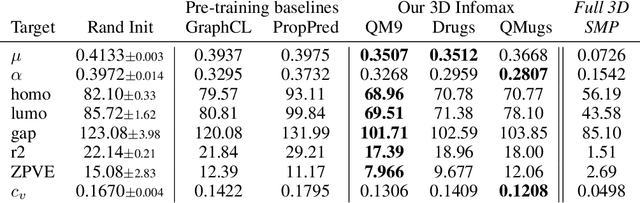
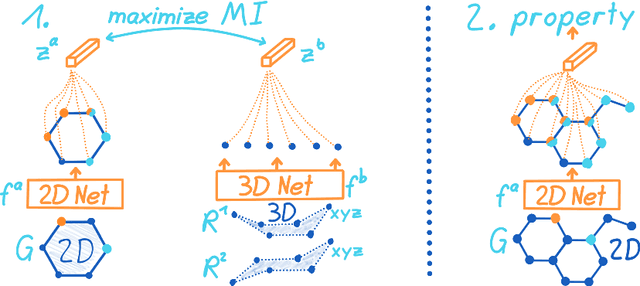

Molecular property prediction is one of the fastest-growing applications of deep learning with critical real-world impacts. Including 3D molecular structure as input to learned models their performance for many molecular tasks. However, this information is infeasible to compute at the scale required by several real-world applications. We propose pre-training a model to reason about the geometry of molecules given only their 2D molecular graphs. Using methods from self-supervised learning, we maximize the mutual information between 3D summary vectors and the representations of a Graph Neural Network (GNN) such that they contain latent 3D information. During fine-tuning on molecules with unknown geometry, the GNN still generates implicit 3D information and can use it to improve downstream tasks. We show that 3D pre-training provides significant improvements for a wide range of properties, such as a 22% average MAE reduction on eight quantum mechanical properties. Moreover, the learned representations can be effectively transferred between datasets in different molecular spaces.
ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High Performance Computing
Jul 20, 2020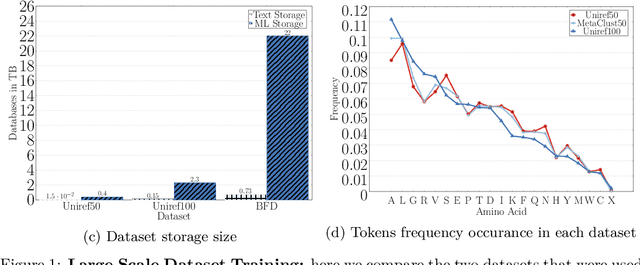
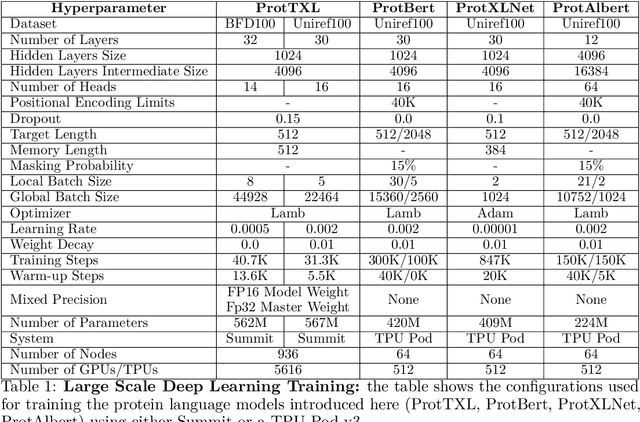
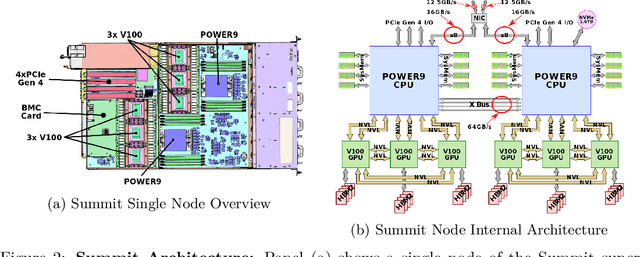
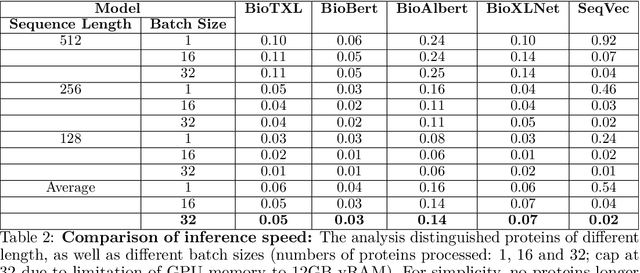
Computational biology and bioinformatics provide vast data gold-mines from protein sequences, ideal for Language Models (LMs) taken from Natural Language Processing (NLP). These LMs reach for new prediction frontiers at low inference costs. Here, we trained two auto-regressive language models (Transformer-XL, XLNet) and two auto-encoder models (Bert, Albert) on data from UniRef and BFD containing up to 393 billion amino acids (words) from 2.1 billion protein sequences (22- and 112-times the entire English Wikipedia). The LMs were trained on the Summit supercomputer at Oak Ridge National Laboratory (ORNL), using 936 nodes (total 5616 GPUs) and one TPU Pod (V3-512 or V3-1024). We validated the advantage of up-scaling LMs to larger models supported by bigger data by predicting secondary structure (3-states: Q3=76-84, 8-states: Q8=65-73), sub-cellular localization for 10 cellular compartments (Q10=74) and whether a protein is membrane-bound or water-soluble (Q2=89). Dimensionality reduction revealed that the LM-embeddings from unlabeled data (only protein sequences) captured important biophysical properties governing protein shape. This implied learning some of the grammar of the language of life realized in protein sequences. The successful up-scaling of protein LMs through HPC to larger data sets slightly reduced the gap between models trained on evolutionary information and LMs. The official GitHub repository: https://github.com/agemagician/ProtTrans