 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge3D Infomax improves GNNs for Molecular Property Prediction
Paper and Code
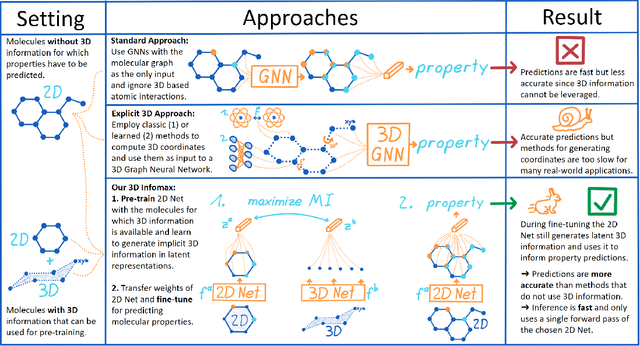
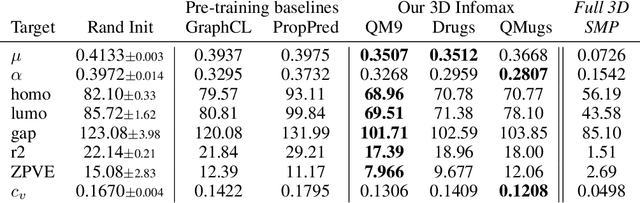
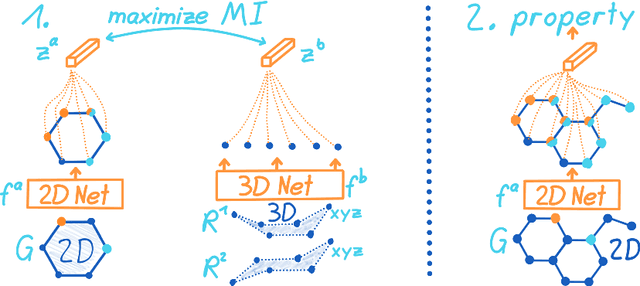

Molecular property prediction is one of the fastest-growing applications of deep learning with critical real-world impacts. Including 3D molecular structure as input to learned models their performance for many molecular tasks. However, this information is infeasible to compute at the scale required by several real-world applications. We propose pre-training a model to reason about the geometry of molecules given only their 2D molecular graphs. Using methods from self-supervised learning, we maximize the mutual information between 3D summary vectors and the representations of a Graph Neural Network (GNN) such that they contain latent 3D information. During fine-tuning on molecules with unknown geometry, the GNN still generates implicit 3D information and can use it to improve downstream tasks. We show that 3D pre-training provides significant improvements for a wide range of properties, such as a 22% average MAE reduction on eight quantum mechanical properties. Moreover, the learned representations can be effectively transferred between datasets in different molecular spaces.