 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTorsional-GFN: a conditional conformation generator for small molecules
Jul 15, 2025Generating stable molecular conformations is crucial in several drug discovery applications, such as estimating the binding affinity of a molecule to a target. Recently, generative machine learning methods have emerged as a promising, more efficient method than molecular dynamics for sampling of conformations from the Boltzmann distribution. In this paper, we introduce Torsional-GFN, a conditional GFlowNet specifically designed to sample conformations of molecules proportionally to their Boltzmann distribution, using only a reward function as training signal. Conditioned on a molecular graph and its local structure (bond lengths and angles), Torsional-GFN samples rotations of its torsion angles. Our results demonstrate that Torsional-GFN is able to sample conformations approximately proportional to the Boltzmann distribution for multiple molecules with a single model, and allows for zero-shot generalization to unseen bond lengths and angles coming from the MD simulations for such molecules. Our work presents a promising avenue for scaling the proposed approach to larger molecular systems, achieving zero-shot generalization to unseen molecules, and including the generation of the local structure into the GFlowNet model.
Virtual Cells: Predict, Explain, Discover
May 20, 2025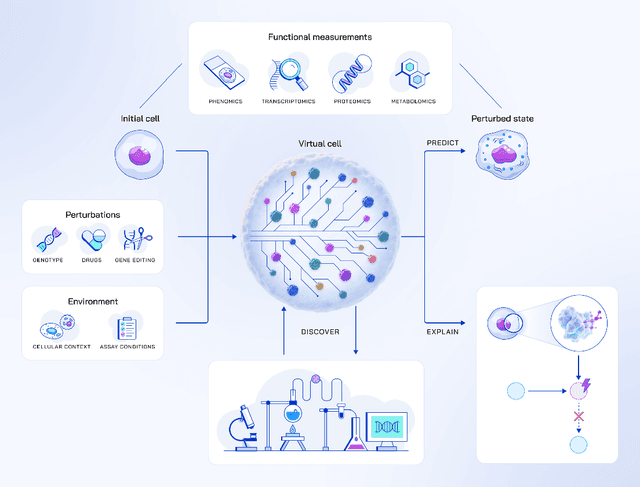



Drug discovery is fundamentally a process of inferring the effects of treatments on patients, and would therefore benefit immensely from computational models that can reliably simulate patient responses, enabling researchers to generate and test large numbers of therapeutic hypotheses safely and economically before initiating costly clinical trials. Even a more specific model that predicts the functional response of cells to a wide range of perturbations would be tremendously valuable for discovering safe and effective treatments that successfully translate to the clinic. Creating such virtual cells has long been a goal of the computational research community that unfortunately remains unachieved given the daunting complexity and scale of cellular biology. Nevertheless, recent advances in AI, computing power, lab automation, and high-throughput cellular profiling provide new opportunities for reaching this goal. In this perspective, we present a vision for developing and evaluating virtual cells that builds on our experience at Recursion. We argue that in order to be a useful tool to discover novel biology, virtual cells must accurately predict the functional response of a cell to perturbations and explain how the predicted response is a consequence of modifications to key biomolecular interactions. We then introduce key principles for designing therapeutically-relevant virtual cells, describe a lab-in-the-loop approach for generating novel insights with them, and advocate for biologically-grounded benchmarks to guide virtual cell development. Finally, we make the case that our approach to virtual cells provides a useful framework for building other models at higher levels of organization, including virtual patients. We hope that these directions prove useful to the research community in developing virtual models optimized for positive impact on drug discovery outcomes.
Implicit Delta Learning of High Fidelity Neural Network Potentials
Dec 08, 2024Neural network potentials (NNPs) offer a fast and accurate alternative to ab-initio methods for molecular dynamics (MD) simulations but are hindered by the high cost of training data from high-fidelity Quantum Mechanics (QM) methods. Our work introduces the Implicit Delta Learning (IDLe) method, which reduces the need for high-fidelity QM data by leveraging cheaper semi-empirical QM computations without compromising NNP accuracy or inference cost. IDLe employs an end-to-end multi-task architecture with fidelity-specific heads that decode energies based on a shared latent representation of the input atomistic system. In various settings, IDLe achieves the same accuracy as single high-fidelity baselines while using up to 50x less high-fidelity data. This result could significantly reduce data generation cost and consequently enhance accuracy and generalization, and expand chemical coverage for NNPs, advancing MD simulations for material science and drug discovery. Additionally, we provide a novel set of 11 million semi-empirical QM calculations to support future multi-fidelity NNP modeling.
OpenQDC: Open Quantum Data Commons
Nov 29, 2024



Machine Learning Interatomic Potentials (MLIPs) are a highly promising alternative to force-fields for molecular dynamics (MD) simulations, offering precise and rapid energy and force calculations. However, Quantum-Mechanical (QM) datasets, crucial for MLIPs, are fragmented across various repositories, hindering accessibility and model development. We introduce the openQDC package, consolidating 37 QM datasets from over 250 quantum methods and 400 million geometries into a single, accessible resource. These datasets are meticulously preprocessed, and standardized for MLIP training, covering a wide range of chemical elements and interactions relevant in organic chemistry. OpenQDC includes tools for normalization and integration, easily accessible via Python. Experiments with well-known architectures like SchNet, TorchMD-Net, and DimeNet reveal challenges for those architectures and constitute a leaderboard to accelerate benchmarking and guide novel algorithms development. Continuously adding datasets to OpenQDC will democratize QM dataset access, foster more collaboration and innovation, enhance MLIP development, and support their adoption in the MD field.
Role of Structural and Conformational Diversity for Machine Learning Potentials
Oct 30, 2023



In the field of Machine Learning Interatomic Potentials (MLIPs), understanding the intricate relationship between data biases, specifically conformational and structural diversity, and model generalization is critical in improving the quality of Quantum Mechanics (QM) data generation efforts. We investigate these dynamics through two distinct experiments: a fixed budget one, where the dataset size remains constant, and a fixed molecular set one, which focuses on fixed structural diversity while varying conformational diversity. Our results reveal nuanced patterns in generalization metrics. Notably, for optimal structural and conformational generalization, a careful balance between structural and conformational diversity is required, but existing QM datasets do not meet that trade-off. Additionally, our results highlight the limitation of the MLIP models at generalizing beyond their training distribution, emphasizing the importance of defining applicability domain during model deployment. These findings provide valuable insights and guidelines for QM data generation efforts.
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
Gotta be SAFE: A New Framework for Molecular Design
Oct 16, 2023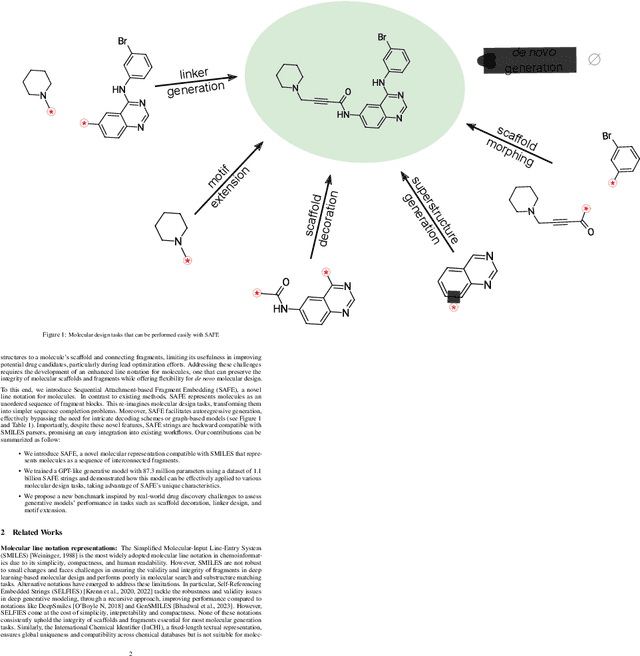

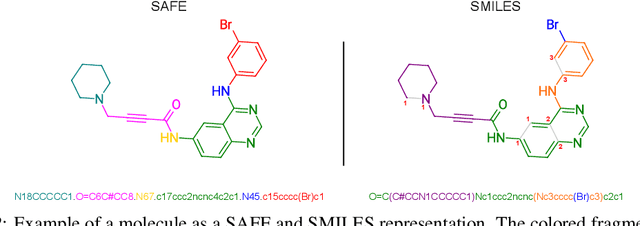
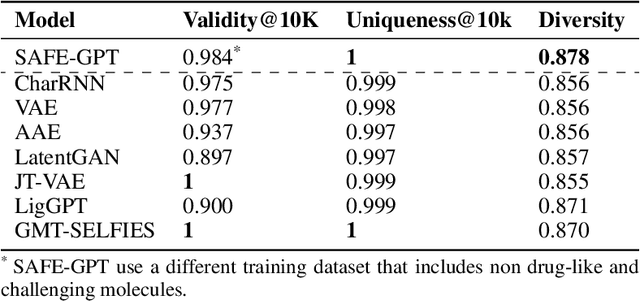
Traditional molecular string representations, such as SMILES, often pose challenges for AI-driven molecular design due to their non-sequential depiction of molecular substructures. To address this issue, we introduce Sequential Attachment-based Fragment Embedding (SAFE), a novel line notation for chemical structures. SAFE reimagines SMILES strings as an unordered sequence of interconnected fragment blocks while maintaining full compatibility with existing SMILES parsers. It streamlines complex generative tasks, including scaffold decoration, fragment linking, polymer generation, and scaffold hopping, while facilitating autoregressive generation for fragment-constrained design, thereby eliminating the need for intricate decoding or graph-based models. We demonstrate the effectiveness of SAFE by training an 87-million-parameter GPT2-like model on a dataset containing 1.1 billion SAFE representations. Through extensive experimentation, we show that our SAFE-GPT model exhibits versatile and robust optimization performance. SAFE opens up new avenues for the rapid exploration of chemical space under various constraints, promising breakthroughs in AI-driven molecular design.
3D Infomax improves GNNs for Molecular Property Prediction
Oct 08, 2021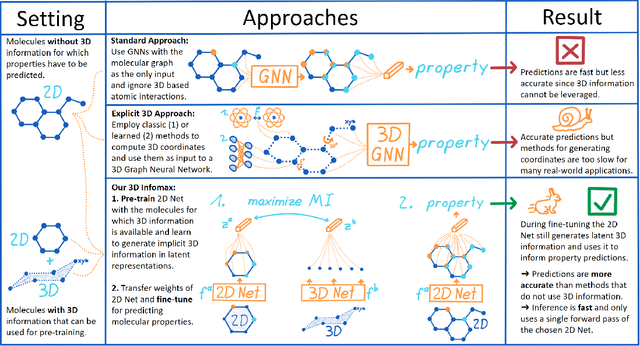
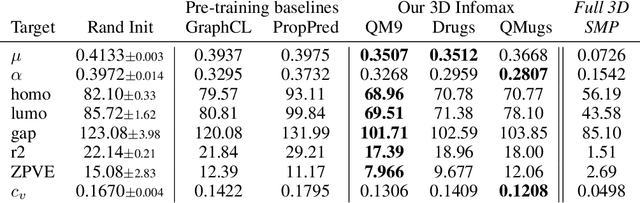
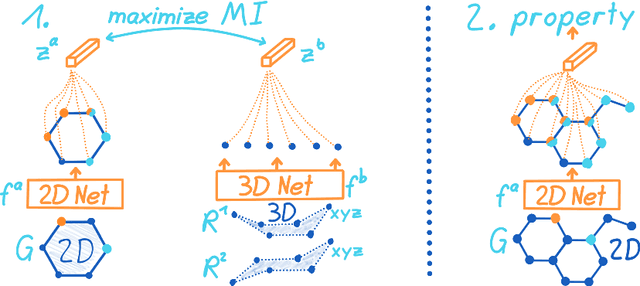

Molecular property prediction is one of the fastest-growing applications of deep learning with critical real-world impacts. Including 3D molecular structure as input to learned models their performance for many molecular tasks. However, this information is infeasible to compute at the scale required by several real-world applications. We propose pre-training a model to reason about the geometry of molecules given only their 2D molecular graphs. Using methods from self-supervised learning, we maximize the mutual information between 3D summary vectors and the representations of a Graph Neural Network (GNN) such that they contain latent 3D information. During fine-tuning on molecules with unknown geometry, the GNN still generates implicit 3D information and can use it to improve downstream tasks. We show that 3D pre-training provides significant improvements for a wide range of properties, such as a 22% average MAE reduction on eight quantum mechanical properties. Moreover, the learned representations can be effectively transferred between datasets in different molecular spaces.
Rethinking Graph Transformers with Spectral Attention
Jun 09, 2021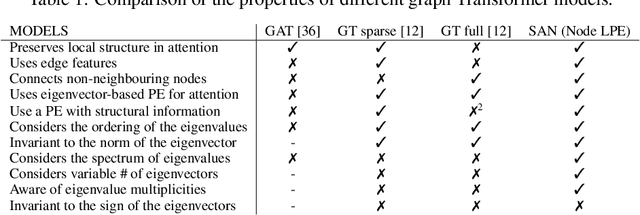
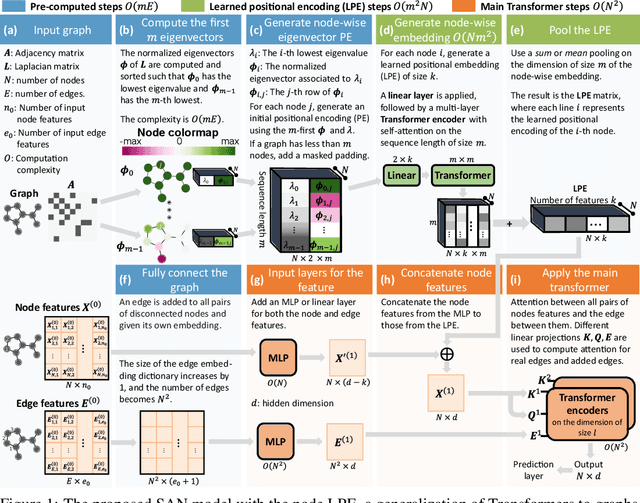

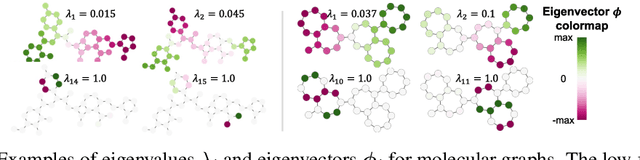
In recent years, the Transformer architecture has proven to be very successful in sequence processing, but its application to other data structures, such as graphs, has remained limited due to the difficulty of properly defining positions. Here, we present the $\textit{Spectral Attention Network}$ (SAN), which uses a learned positional encoding (LPE) that can take advantage of the full Laplacian spectrum to learn the position of each node in a given graph. This LPE is then added to the node features of the graph and passed to a fully-connected Transformer. By leveraging the full spectrum of the Laplacian, our model is theoretically powerful in distinguishing graphs, and can better detect similar sub-structures from their resonance. Further, by fully connecting the graph, the Transformer does not suffer from over-squashing, an information bottleneck of most GNNs, and enables better modeling of physical phenomenons such as heat transfer and electric interaction. When tested empirically on a set of 4 standard datasets, our model performs on par or better than state-of-the-art GNNs, and outperforms any attention-based model by a wide margin, becoming the first fully-connected architecture to perform well on graph benchmarks.
Geodesics in fibered latent spaces: A geometric approach to learning correspondences between conditions
May 24, 2020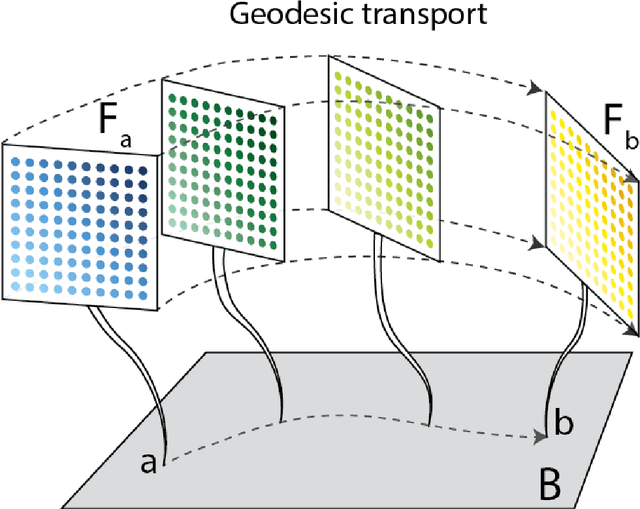
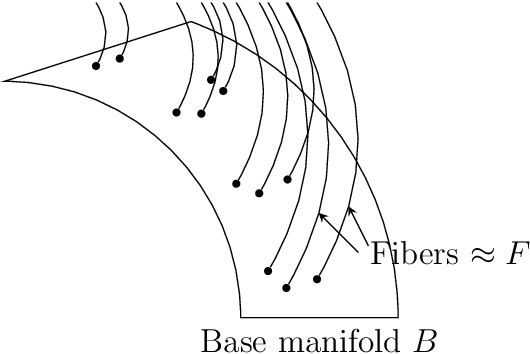
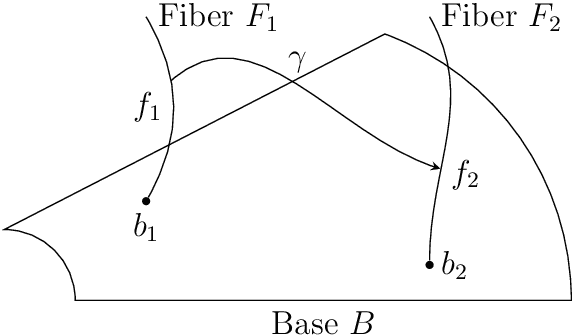
This work introduces a geometric framework and a novel network architecture for creating correspondences between samples of different conditions. Under this formalism, the latent space is a fiber bundle stratified into a base space encoding conditions, and a fiber space encoding the variations within conditions. The correspondences between conditions are obtained by minimizing an energy functional, resulting in diffeomorphism flows between fibers. We illustrate this approach using MNIST and Olivetti and benchmark its performances on the task of batch correction, which is the problem of integrating multiple biological datasets together.