 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImplicit Delta Learning of High Fidelity Neural Network Potentials
Dec 08, 2024Neural network potentials (NNPs) offer a fast and accurate alternative to ab-initio methods for molecular dynamics (MD) simulations but are hindered by the high cost of training data from high-fidelity Quantum Mechanics (QM) methods. Our work introduces the Implicit Delta Learning (IDLe) method, which reduces the need for high-fidelity QM data by leveraging cheaper semi-empirical QM computations without compromising NNP accuracy or inference cost. IDLe employs an end-to-end multi-task architecture with fidelity-specific heads that decode energies based on a shared latent representation of the input atomistic system. In various settings, IDLe achieves the same accuracy as single high-fidelity baselines while using up to 50x less high-fidelity data. This result could significantly reduce data generation cost and consequently enhance accuracy and generalization, and expand chemical coverage for NNPs, advancing MD simulations for material science and drug discovery. Additionally, we provide a novel set of 11 million semi-empirical QM calculations to support future multi-fidelity NNP modeling.
OpenQDC: Open Quantum Data Commons
Nov 29, 2024



Machine Learning Interatomic Potentials (MLIPs) are a highly promising alternative to force-fields for molecular dynamics (MD) simulations, offering precise and rapid energy and force calculations. However, Quantum-Mechanical (QM) datasets, crucial for MLIPs, are fragmented across various repositories, hindering accessibility and model development. We introduce the openQDC package, consolidating 37 QM datasets from over 250 quantum methods and 400 million geometries into a single, accessible resource. These datasets are meticulously preprocessed, and standardized for MLIP training, covering a wide range of chemical elements and interactions relevant in organic chemistry. OpenQDC includes tools for normalization and integration, easily accessible via Python. Experiments with well-known architectures like SchNet, TorchMD-Net, and DimeNet reveal challenges for those architectures and constitute a leaderboard to accelerate benchmarking and guide novel algorithms development. Continuously adding datasets to OpenQDC will democratize QM dataset access, foster more collaboration and innovation, enhance MLIP development, and support their adoption in the MD field.
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
Gotta be SAFE: A New Framework for Molecular Design
Oct 16, 2023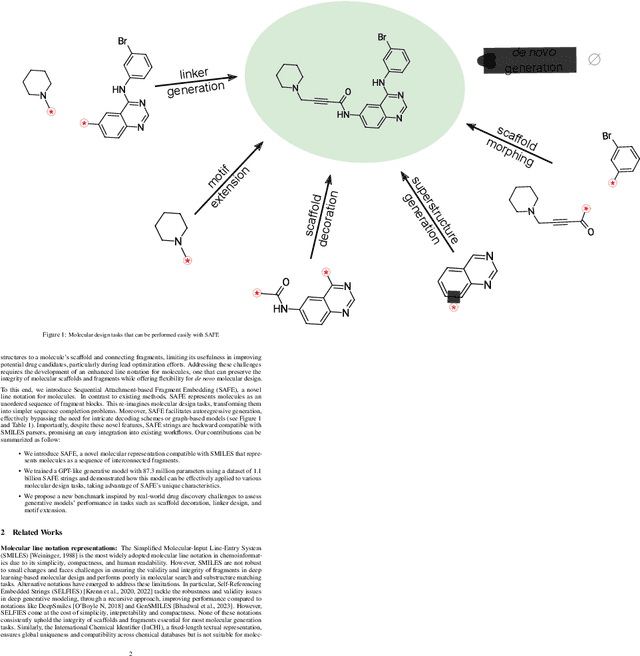

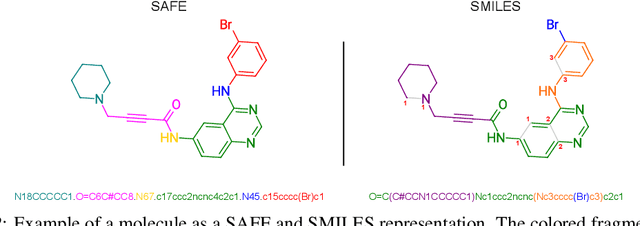
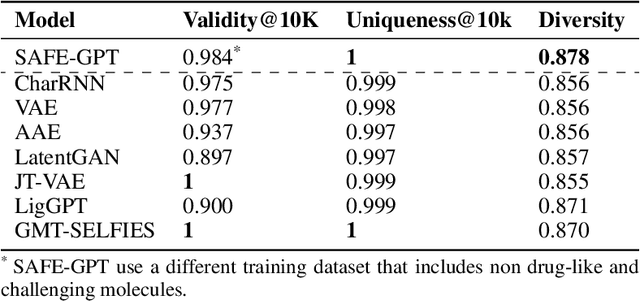
Traditional molecular string representations, such as SMILES, often pose challenges for AI-driven molecular design due to their non-sequential depiction of molecular substructures. To address this issue, we introduce Sequential Attachment-based Fragment Embedding (SAFE), a novel line notation for chemical structures. SAFE reimagines SMILES strings as an unordered sequence of interconnected fragment blocks while maintaining full compatibility with existing SMILES parsers. It streamlines complex generative tasks, including scaffold decoration, fragment linking, polymer generation, and scaffold hopping, while facilitating autoregressive generation for fragment-constrained design, thereby eliminating the need for intricate decoding or graph-based models. We demonstrate the effectiveness of SAFE by training an 87-million-parameter GPT2-like model on a dataset containing 1.1 billion SAFE representations. Through extensive experimentation, we show that our SAFE-GPT model exhibits versatile and robust optimization performance. SAFE opens up new avenues for the rapid exploration of chemical space under various constraints, promising breakthroughs in AI-driven molecular design.