 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeElucidating the Design Space of Flow Matching for Cellular Microscopy
Mar 25, 2026Flow-matching generative models are increasingly used to simulate cell responses to biological perturbations. However, the design space for building such models is large and underexplored. We systematically analyse the design space of flow matching models for cell-microscopy images, finding that many popular techniques are unnecessary and can even hurt performance. We develop a simple, stable, and scalable recipe which we use to train our foundation model. We scale our model to two orders of magnitude larger than prior methods, achieving a two-fold FID and ten-fold KID improvement over prior methods. We then fine-tune our model with pre-trained molecular embeddings to achieve state-of-the-art performance simulating responses to unseen molecules. Code is available at https://github.com/valence-labs/microscopy-flow-matching
A Cross Modal Knowledge Distillation & Data Augmentation Recipe for Improving Transcriptomics Representations through Morphological Features
May 27, 2025Understanding cellular responses to stimuli is crucial for biological discovery and drug development. Transcriptomics provides interpretable, gene-level insights, while microscopy imaging offers rich predictive features but is harder to interpret. Weakly paired datasets, where samples share biological states, enable multimodal learning but are scarce, limiting their utility for training and multimodal inference. We propose a framework to enhance transcriptomics by distilling knowledge from microscopy images. Using weakly paired data, our method aligns and binds modalities, enriching gene expression representations with morphological information. To address data scarcity, we introduce (1) Semi-Clipped, an adaptation of CLIP for cross-modal distillation using pretrained foundation models, achieving state-of-the-art results, and (2) PEA (Perturbation Embedding Augmentation), a novel augmentation technique that enhances transcriptomics data while preserving inherent biological information. These strategies improve the predictive power and retain the interpretability of transcriptomics, enabling rich unimodal representations for complex biological tasks.
TxPert: Leveraging Biochemical Relationships for Out-of-Distribution Transcriptomic Perturbation Prediction
May 20, 2025Accurately predicting cellular responses to genetic perturbations is essential for understanding disease mechanisms and designing effective therapies. Yet exhaustively exploring the space of possible perturbations (e.g., multi-gene perturbations or across tissues and cell types) is prohibitively expensive, motivating methods that can generalize to unseen conditions. In this work, we explore how knowledge graphs of gene-gene relationships can improve out-of-distribution (OOD) prediction across three challenging settings: unseen single perturbations; unseen double perturbations; and unseen cell lines. In particular, we present: (i) TxPert, a new state-of-the-art method that leverages multiple biological knowledge networks to predict transcriptional responses under OOD scenarios; (ii) an in-depth analysis demonstrating the impact of graphs, model architecture, and data on performance; and (iii) an expanded benchmarking framework that strengthens evaluation standards for perturbation modeling.
Virtual Cells: Predict, Explain, Discover
May 20, 2025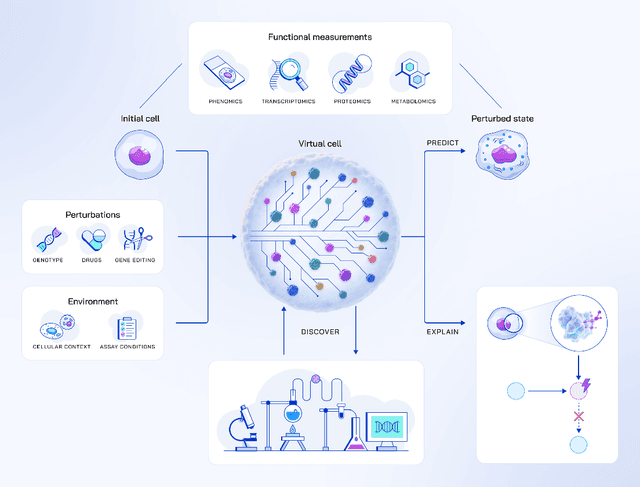



Drug discovery is fundamentally a process of inferring the effects of treatments on patients, and would therefore benefit immensely from computational models that can reliably simulate patient responses, enabling researchers to generate and test large numbers of therapeutic hypotheses safely and economically before initiating costly clinical trials. Even a more specific model that predicts the functional response of cells to a wide range of perturbations would be tremendously valuable for discovering safe and effective treatments that successfully translate to the clinic. Creating such virtual cells has long been a goal of the computational research community that unfortunately remains unachieved given the daunting complexity and scale of cellular biology. Nevertheless, recent advances in AI, computing power, lab automation, and high-throughput cellular profiling provide new opportunities for reaching this goal. In this perspective, we present a vision for developing and evaluating virtual cells that builds on our experience at Recursion. We argue that in order to be a useful tool to discover novel biology, virtual cells must accurately predict the functional response of a cell to perturbations and explain how the predicted response is a consequence of modifications to key biomolecular interactions. We then introduce key principles for designing therapeutically-relevant virtual cells, describe a lab-in-the-loop approach for generating novel insights with them, and advocate for biologically-grounded benchmarks to guide virtual cell development. Finally, we make the case that our approach to virtual cells provides a useful framework for building other models at higher levels of organization, including virtual patients. We hope that these directions prove useful to the research community in developing virtual models optimized for positive impact on drug discovery outcomes.
Graph and Simplicial Complex Prediction Gaussian Process via the Hodgelet Representations
May 16, 2025Predicting the labels of graph-structured data is crucial in scientific applications and is often achieved using graph neural networks (GNNs). However, when data is scarce, GNNs suffer from overfitting, leading to poor performance. Recently, Gaussian processes (GPs) with graph-level inputs have been proposed as an alternative. In this work, we extend the Gaussian process framework to simplicial complexes (SCs), enabling the handling of edge-level attributes and attributes supported on higher-order simplices. We further augment the resulting SC representations by considering their Hodge decompositions, allowing us to account for homological information, such as the number of holes, in the SC. We demonstrate that our framework enhances the predictions across various applications, paving the way for GPs to be more widely used for graph and SC-level predictions.
SAFE setup for generative molecular design
Oct 26, 2024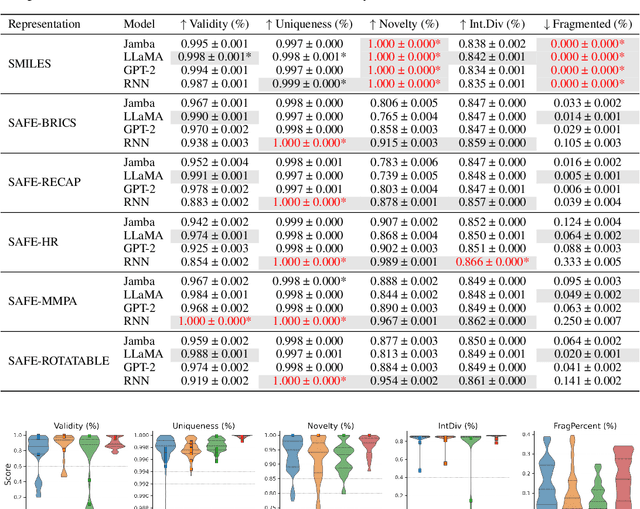
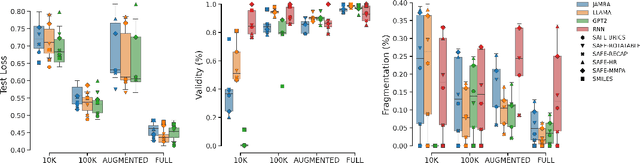
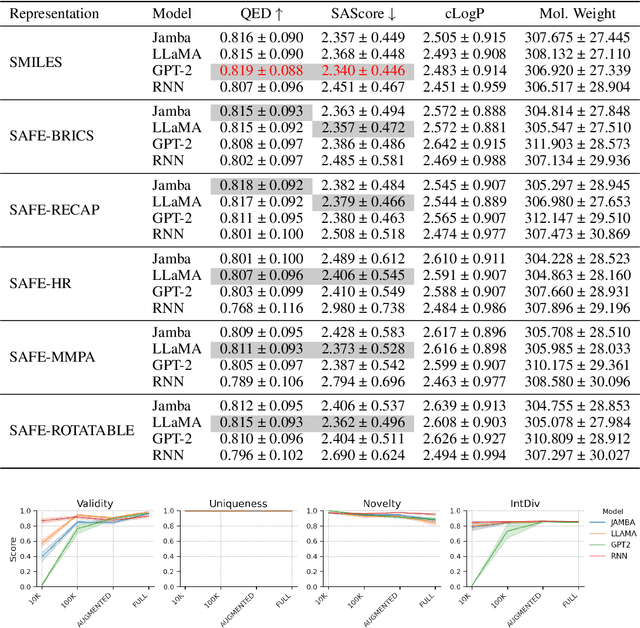
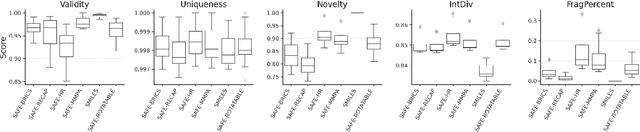
SMILES-based molecular generative models have been pivotal in drug design but face challenges in fragment-constrained tasks. To address this, the Sequential Attachment-based Fragment Embedding (SAFE) representation was recently introduced as an alternative that streamlines those tasks. In this study, we investigate the optimal setups for training SAFE generative models, focusing on dataset size, data augmentation through randomization, model architecture, and bond disconnection algorithms. We found that larger, more diverse datasets improve performance, with the LLaMA architecture using Rotary Positional Embedding proving most robust. SAFE-based models also consistently outperform SMILES-based approaches in scaffold decoration and linker design, particularly with BRICS decomposition yielding the best results. These insights highlight key factors that significantly impact the efficacy of SAFE-based generative models.
Benchmarking Transcriptomics Foundation Models for Perturbation Analysis : one PCA still rules them all
Oct 17, 2024
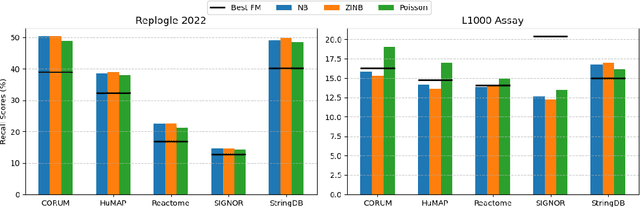
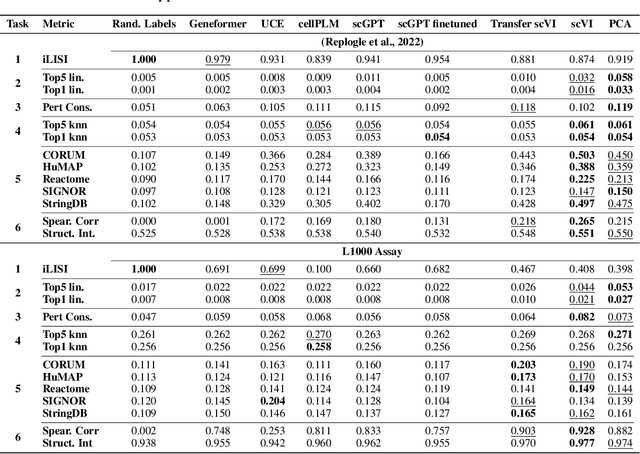
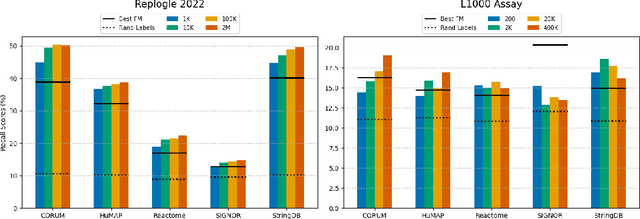
Understanding the relationships among genes, compounds, and their interactions in living organisms remains limited due to technological constraints and the complexity of biological data. Deep learning has shown promise in exploring these relationships using various data types. However, transcriptomics, which provides detailed insights into cellular states, is still underused due to its high noise levels and limited data availability. Recent advancements in transcriptomics sequencing provide new opportunities to uncover valuable insights, especially with the rise of many new foundation models for transcriptomics, yet no benchmark has been made to robustly evaluate the effectiveness of these rising models for perturbation analysis. This article presents a novel biologically motivated evaluation framework and a hierarchy of perturbation analysis tasks for comparing the performance of pretrained foundation models to each other and to more classical techniques of learning from transcriptomics data. We compile diverse public datasets from different sequencing techniques and cell lines to assess models performance. Our approach identifies scVI and PCA to be far better suited models for understanding biological perturbations in comparison to existing foundation models, especially in their application in real-world scenarios.
Role of Structural and Conformational Diversity for Machine Learning Potentials
Oct 30, 2023



In the field of Machine Learning Interatomic Potentials (MLIPs), understanding the intricate relationship between data biases, specifically conformational and structural diversity, and model generalization is critical in improving the quality of Quantum Mechanics (QM) data generation efforts. We investigate these dynamics through two distinct experiments: a fixed budget one, where the dataset size remains constant, and a fixed molecular set one, which focuses on fixed structural diversity while varying conformational diversity. Our results reveal nuanced patterns in generalization metrics. Notably, for optimal structural and conformational generalization, a careful balance between structural and conformational diversity is required, but existing QM datasets do not meet that trade-off. Additionally, our results highlight the limitation of the MLIP models at generalizing beyond their training distribution, emphasizing the importance of defining applicability domain during model deployment. These findings provide valuable insights and guidelines for QM data generation efforts.
Gotta be SAFE: A New Framework for Molecular Design
Oct 16, 2023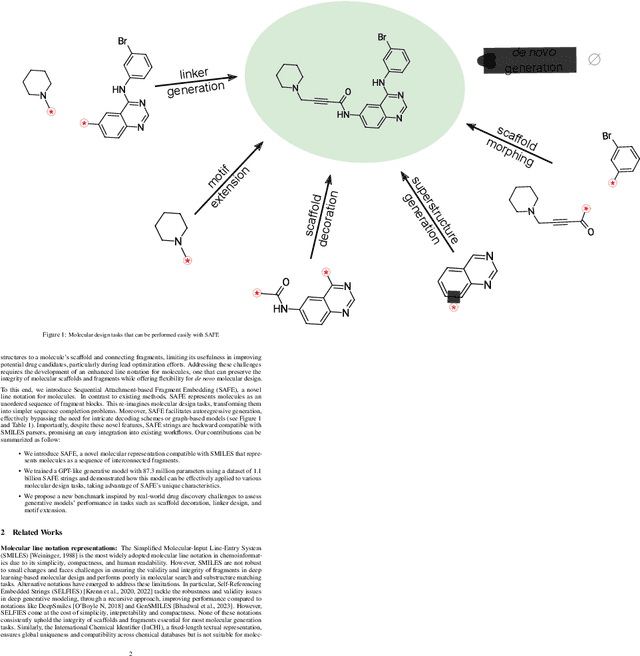

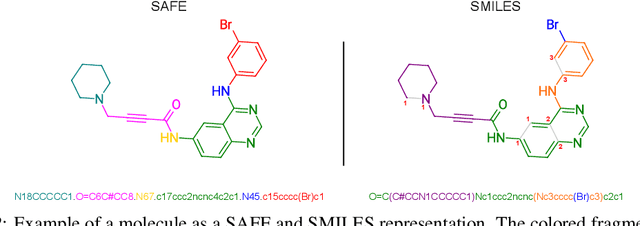
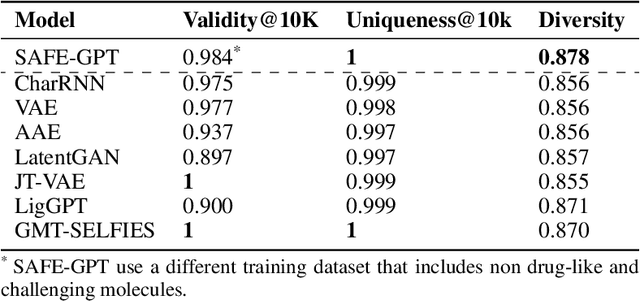
Traditional molecular string representations, such as SMILES, often pose challenges for AI-driven molecular design due to their non-sequential depiction of molecular substructures. To address this issue, we introduce Sequential Attachment-based Fragment Embedding (SAFE), a novel line notation for chemical structures. SAFE reimagines SMILES strings as an unordered sequence of interconnected fragment blocks while maintaining full compatibility with existing SMILES parsers. It streamlines complex generative tasks, including scaffold decoration, fragment linking, polymer generation, and scaffold hopping, while facilitating autoregressive generation for fragment-constrained design, thereby eliminating the need for intricate decoding or graph-based models. We demonstrate the effectiveness of SAFE by training an 87-million-parameter GPT2-like model on a dataset containing 1.1 billion SAFE representations. Through extensive experimentation, we show that our SAFE-GPT model exhibits versatile and robust optimization performance. SAFE opens up new avenues for the rapid exploration of chemical space under various constraints, promising breakthroughs in AI-driven molecular design.
Molecular Design in Synthetically Accessible Chemical Space via Deep Reinforcement Learning
Apr 29, 2020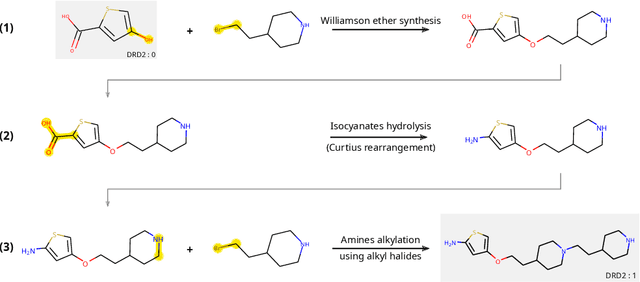
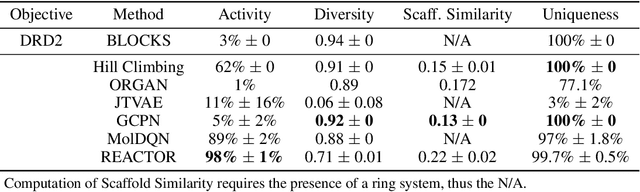
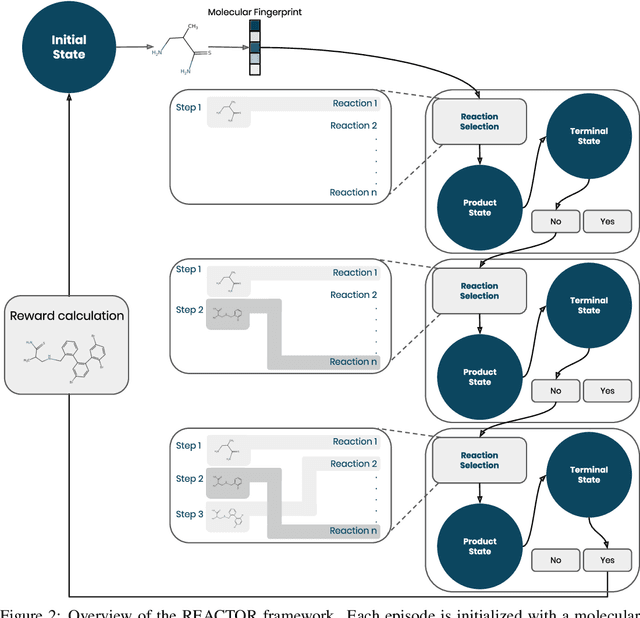
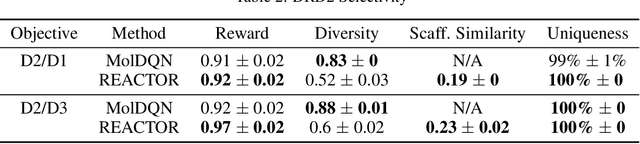
The fundamental goal of generative drug design is to propose optimized molecules that meet predefined activity, selectivity, and pharmacokinetic criteria. Despite recent progress, we argue that existing generative methods are limited in their ability to favourably shift the distributions of molecular properties during optimization. We instead propose a novel Reinforcement Learning framework for molecular design in which an agent learns to directly optimize through a space of synthetically-accessible drug-like molecules. This becomes possible by defining transitions in our Markov Decision Process as chemical reactions, and allows us to leverage synthetic routes as an inductive bias. We validate our method by demonstrating that it outperforms existing state-of the art approaches in the optimization of pharmacologically-relevant objectives, while results on multi-objective optimization tasks suggest increased scalability to realistic pharmaceutical design problems.