 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeElucidating the Design Space of Flow Matching for Cellular Microscopy
Mar 25, 2026Flow-matching generative models are increasingly used to simulate cell responses to biological perturbations. However, the design space for building such models is large and underexplored. We systematically analyse the design space of flow matching models for cell-microscopy images, finding that many popular techniques are unnecessary and can even hurt performance. We develop a simple, stable, and scalable recipe which we use to train our foundation model. We scale our model to two orders of magnitude larger than prior methods, achieving a two-fold FID and ten-fold KID improvement over prior methods. We then fine-tune our model with pre-trained molecular embeddings to achieve state-of-the-art performance simulating responses to unseen molecules. Code is available at https://github.com/valence-labs/microscopy-flow-matching
Virtual Cells: Predict, Explain, Discover
May 20, 2025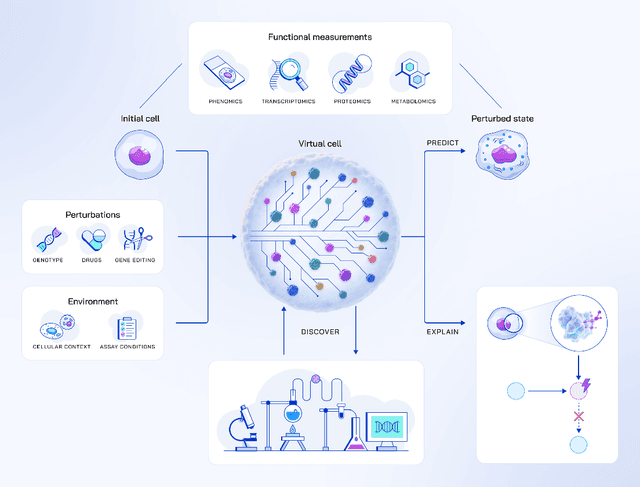



Drug discovery is fundamentally a process of inferring the effects of treatments on patients, and would therefore benefit immensely from computational models that can reliably simulate patient responses, enabling researchers to generate and test large numbers of therapeutic hypotheses safely and economically before initiating costly clinical trials. Even a more specific model that predicts the functional response of cells to a wide range of perturbations would be tremendously valuable for discovering safe and effective treatments that successfully translate to the clinic. Creating such virtual cells has long been a goal of the computational research community that unfortunately remains unachieved given the daunting complexity and scale of cellular biology. Nevertheless, recent advances in AI, computing power, lab automation, and high-throughput cellular profiling provide new opportunities for reaching this goal. In this perspective, we present a vision for developing and evaluating virtual cells that builds on our experience at Recursion. We argue that in order to be a useful tool to discover novel biology, virtual cells must accurately predict the functional response of a cell to perturbations and explain how the predicted response is a consequence of modifications to key biomolecular interactions. We then introduce key principles for designing therapeutically-relevant virtual cells, describe a lab-in-the-loop approach for generating novel insights with them, and advocate for biologically-grounded benchmarks to guide virtual cell development. Finally, we make the case that our approach to virtual cells provides a useful framework for building other models at higher levels of organization, including virtual patients. We hope that these directions prove useful to the research community in developing virtual models optimized for positive impact on drug discovery outcomes.
Towards scientific discovery with dictionary learning: Extracting biological concepts from microscopy foundation models
Dec 20, 2024Dictionary learning (DL) has emerged as a powerful interpretability tool for large language models. By extracting known concepts (e.g., Golden-Gate Bridge) from human-interpretable data (e.g., text), sparse DL can elucidate a model's inner workings. In this work, we ask if DL can also be used to discover unknown concepts from less human-interpretable scientific data (e.g., cell images), ultimately enabling modern approaches to scientific discovery. As a first step, we use DL algorithms to study microscopy foundation models trained on multi-cell image data, where little prior knowledge exists regarding which high-level concepts should arise. We show that sparse dictionaries indeed extract biologically-meaningful concepts such as cell type and genetic perturbation type. We also propose a new DL algorithm, Iterative Codebook Feature Learning~(ICFL), and combine it with a pre-processing step that uses PCA whitening from a control dataset. In our experiments, we demonstrate that both ICFL and PCA improve the selectivity of extracted features compared to TopK sparse autoencoders.
ViTally Consistent: Scaling Biological Representation Learning for Cell Microscopy
Nov 04, 2024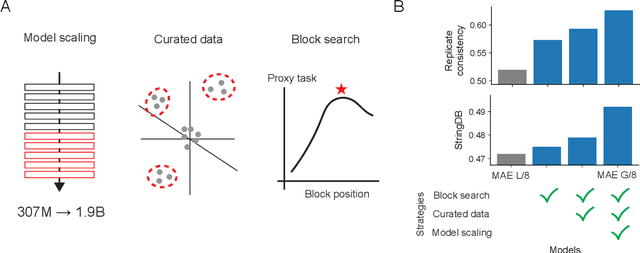
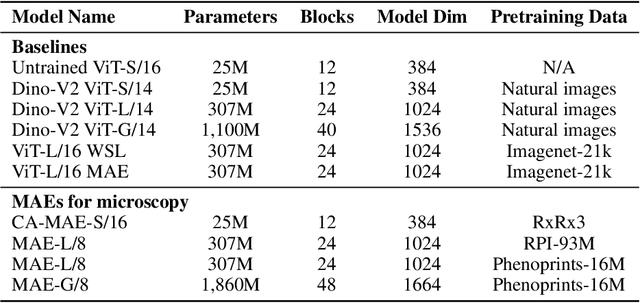
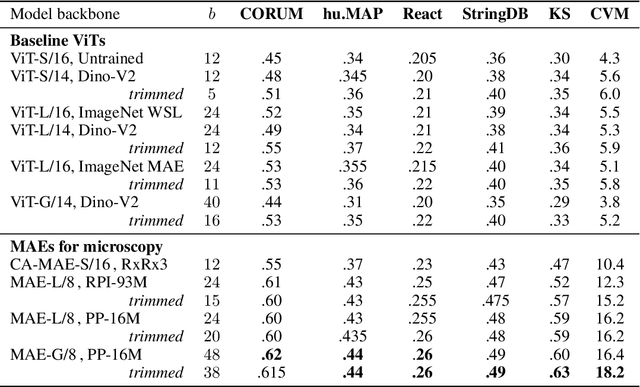
Large-scale cell microscopy screens are used in drug discovery and molecular biology research to study the effects of millions of chemical and genetic perturbations on cells. To use these images in downstream analysis, we need models that can map each image into a feature space that represents diverse biological phenotypes consistently, in the sense that perturbations with similar biological effects have similar representations. In this work, we present the largest foundation model for cell microscopy data to date, a new 1.9 billion-parameter ViT-G/8 MAE trained on over 8 billion microscopy image crops. Compared to a previous published ViT-L/8 MAE, our new model achieves a 60% improvement in linear separability of genetic perturbations and obtains the best overall performance on whole-genome biological relationship recall and replicate consistency benchmarks. Beyond scaling, we developed two key methods that improve performance: (1) training on a curated and diverse dataset; and, (2) using biologically motivated linear probing tasks to search across each transformer block for the best candidate representation of whole-genome screens. We find that many self-supervised vision transformers, pretrained on either natural or microscopy images, yield significantly more biologically meaningful representations of microscopy images in their intermediate blocks than in their typically used final blocks. More broadly, our approach and results provide insights toward a general strategy for successfully building foundation models for large-scale biological data.
Efficient Biological Data Acquisition through Inference Set Design
Oct 25, 2024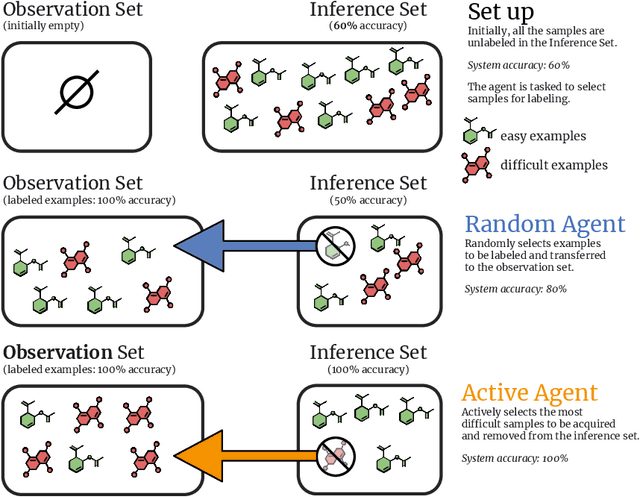

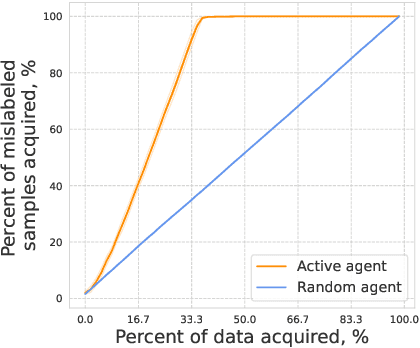

In drug discovery, highly automated high-throughput laboratories are used to screen a large number of compounds in search of effective drugs. These experiments are expensive, so we might hope to reduce their cost by experimenting on a subset of the compounds, and predicting the outcomes of the remaining experiments. In this work, we model this scenario as a sequential subset selection problem: we aim to select the smallest set of candidates in order to achieve some desired level of accuracy for the system as a whole. Our key observation is that, if there is heterogeneity in the difficulty of the prediction problem across the input space, selectively obtaining the labels for the hardest examples in the acquisition pool will leave only the relatively easy examples to remain in the inference set, leading to better overall system performance. We call this mechanism inference set design, and propose the use of an uncertainty-based active learning solution to prune out these challenging examples. Our algorithm includes an explicit stopping criterion that stops running the experiments when it is sufficiently confident that the system has reached the target performance. Our empirical studies on image and molecular datasets, as well as a real-world large-scale biological assay, show that deploying active learning for inference set design leads to significant reduction in experimental cost while obtaining high system performance.
Automated Discovery of Pairwise Interactions from Unstructured Data
Sep 11, 2024



Pairwise interactions between perturbations to a system can provide evidence for the causal dependencies of the underlying underlying mechanisms of a system. When observations are low dimensional, hand crafted measurements, detecting interactions amounts to simple statistical tests, but it is not obvious how to detect interactions between perturbations affecting latent variables. We derive two interaction tests that are based on pairwise interventions, and show how these tests can be integrated into an active learning pipeline to efficiently discover pairwise interactions between perturbations. We illustrate the value of these tests in the context of biology, where pairwise perturbation experiments are frequently used to reveal interactions that are not observable from any single perturbation. Our tests can be run on unstructured data, such as the pixels in an image, which enables a more general notion of interaction than typical cell viability experiments, and can be run on cheaper experimental assays. We validate on several synthetic and real biological experiments that our tests are able to identify interacting pairs effectively. We evaluate our approach on a real biological experiment where we knocked out 50 pairs of genes and measured the effect with microscopy images. We show that we are able to recover significantly more known biological interactions than random search and standard active learning baselines.
Sparsity regularization via tree-structured environments for disentangled representations
Jun 10, 2024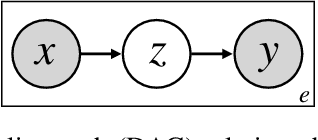

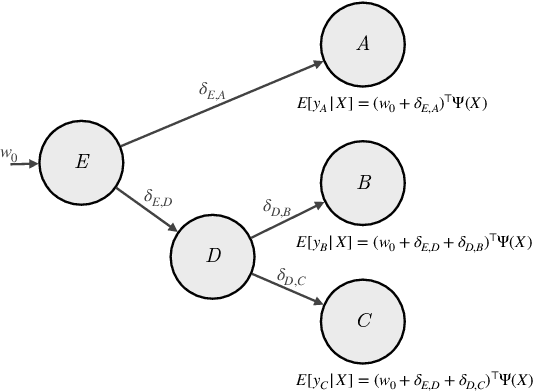
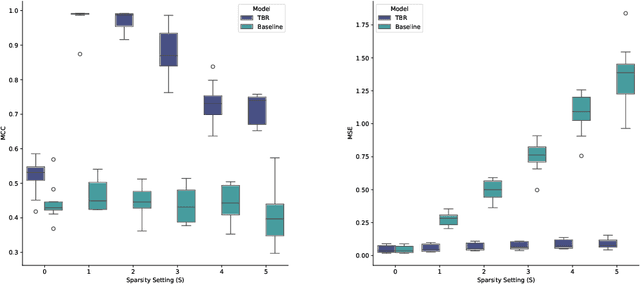
Many causal systems such as biological processes in cells can only be observed indirectly via measurements, such as gene expression. Causal representation learning -- the task of correctly mapping low-level observations to latent causal variables -- could advance scientific understanding by enabling inference of latent variables such as pathway activation. In this paper, we develop methods for inferring latent variables from multiple related datasets (environments) and tasks. As a running example, we consider the task of predicting a phenotype from gene expression, where we often collect data from multiple cell types or organisms that are related in known ways. The key insight is that the mapping from latent variables driven by gene expression to the phenotype of interest changes sparsely across closely related environments. To model sparse changes, we introduce Tree-Based Regularization (TBR), an objective that minimizes both prediction error and regularizes closely related environments to learn similar predictors. We prove that under assumptions about the degree of sparse changes, TBR identifies the true latent variables up to some simple transformations. We evaluate the theory empirically with both simulations and ground-truth gene expression data. We find that TBR recovers the latent causal variables better than related methods across these settings, even under settings that violate some assumptions of the theory.
Targeted Sequential Indirect Experiment Design
May 30, 2024

Scientific hypotheses typically concern specific aspects of complex, imperfectly understood or entirely unknown mechanisms, such as the effect of gene expression levels on phenotypes or how microbial communities influence environmental health. Such queries are inherently causal (rather than purely associational), but in many settings, experiments can not be conducted directly on the target variables of interest, but are indirect. Therefore, they perturb the target variable, but do not remove potential confounding factors. If, additionally, the resulting experimental measurements are multi-dimensional and the studied mechanisms nonlinear, the query of interest is generally not identified. We develop an adaptive strategy to design indirect experiments that optimally inform a targeted query about the ground truth mechanism in terms of sequentially narrowing the gap between an upper and lower bound on the query. While the general formulation consists of a bi-level optimization procedure, we derive an efficiently estimable analytical kernel-based estimator of the bounds for the causal effect, a query of key interest, and demonstrate the efficacy of our approach in confounded, multivariate, nonlinear synthetic settings.
Leveraging Structure Between Environments: Phylogenetic Regularization Incentivizes Disentangled Representations
May 30, 2024Many causal systems such as biological processes in cells can only be observed indirectly via measurements, such as gene expression. Causal representation learning -- the task of correctly mapping low-level observations to latent causal variables -- could advance scientific understanding by enabling inference of latent variables such as pathway activation. In this paper, we develop methods for inferring latent variables from multiple related datasets (environments) and tasks. As a running example, we consider the task of predicting a phenotype from gene expression, where we often collect data from multiple cell types or organisms that are related in known ways. The key insight is that the mapping from latent variables driven by gene expression to the phenotype of interest changes sparsely across closely related environments. To model sparse changes, we introduce Tree-Based Regularization (TBR), an objective that minimizes both prediction error and regularizes closely related environments to learn similar predictors. We prove that under assumptions about the degree of sparse changes, TBR identifies the true latent variables up to some simple transformations. We evaluate the theory empirically with both simulations and ground-truth gene expression data. We find that TBR recovers the latent causal variables better than related methods across these settings, even under settings that violate some assumptions of the theory.
Propensity Score Alignment of Unpaired Multimodal Data
Apr 02, 2024Multimodal representation learning techniques typically rely on paired samples to learn common representations, but paired samples are challenging to collect in fields such as biology where measurement devices often destroy the samples. This paper presents an approach to address the challenge of aligning unpaired samples across disparate modalities in multimodal representation learning. We draw an analogy between potential outcomes in causal inference and potential views in multimodal observations, which allows us to use Rubin's framework to estimate a common space in which to match samples. Our approach assumes we collect samples that are experimentally perturbed by treatments, and uses this to estimate a propensity score from each modality, which encapsulates all shared information between a latent state and treatment and can be used to define a distance between samples. We experiment with two alignment techniques that leverage this distance -- shared nearest neighbours (SNN) and optimal transport (OT) matching -- and find that OT matching results in significant improvements over state-of-the-art alignment approaches in both a synthetic multi-modal setting and in real-world data from NeurIPS Multimodal Single-Cell Integration Challenge.