 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLost in Tokenization: Context as the Key to Unlocking Biomolecular Understanding in Scientific LLMs
Oct 27, 2025Scientific Large Language Models (Sci-LLMs) have emerged as a promising frontier for accelerating biological discovery. However, these models face a fundamental challenge when processing raw biomolecular sequences: the tokenization dilemma. Whether treating sequences as a specialized language, risking the loss of functional motif information, or as a separate modality, introducing formidable alignment challenges, current strategies fundamentally limit their reasoning capacity. We challenge this sequence-centric paradigm by positing that a more effective strategy is to provide Sci-LLMs with high-level structured context derived from established bioinformatics tools, thereby bypassing the need to interpret low-level noisy sequence data directly. Through a systematic comparison of leading Sci-LLMs on biological reasoning tasks, we tested three input modes: sequence-only, context-only, and a combination of both. Our findings are striking: the context-only approach consistently and substantially outperforms all other modes. Even more revealing, the inclusion of the raw sequence alongside its high-level context consistently degrades performance, indicating that raw sequences act as informational noise, even for models with specialized tokenization schemes. These results suggest that the primary strength of existing Sci-LLMs lies not in their nascent ability to interpret biomolecular syntax from scratch, but in their profound capacity for reasoning over structured, human-readable knowledge. Therefore, we argue for reframing Sci-LLMs not as sequence decoders, but as powerful reasoning engines over expert knowledge. This work lays the foundation for a new class of hybrid scientific AI agents, repositioning the developmental focus from direct sequence interpretation towards high-level knowledge synthesis. The code is available at github.com/opendatalab-raise-dev/CoKE.
Predicting function of evolutionarily implausible DNA sequences
Jun 12, 2025Genomic language models (gLMs) show potential for generating novel, functional DNA sequences for synthetic biology, but doing so requires them to learn not just evolutionary plausibility, but also sequence-to-function relationships. We introduce a set of prediction tasks called Nullsettes, which assesses a model's ability to predict loss-of-function mutations created by translocating key control elements in synthetic expression cassettes. Across 12 state-of-the-art models, we find that mutation effect prediction performance strongly correlates with the predicted likelihood of the nonmutant. Furthermore, the range of likelihood values predictive of strong model performance is highly dependent on sequence length. Our work highlights the importance of considering both sequence likelihood and sequence length when using gLMs for mutation effect prediction.
ViTally Consistent: Scaling Biological Representation Learning for Cell Microscopy
Nov 04, 2024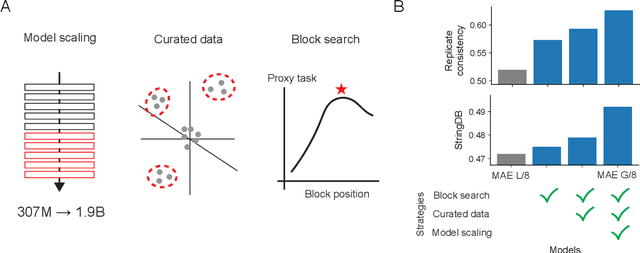
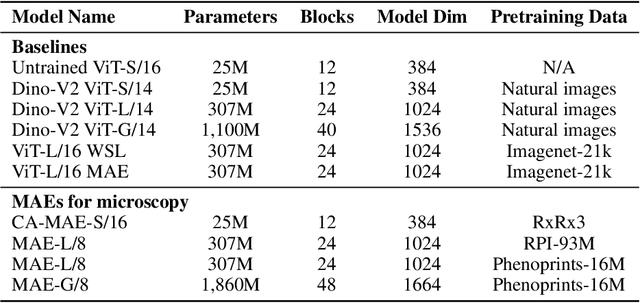
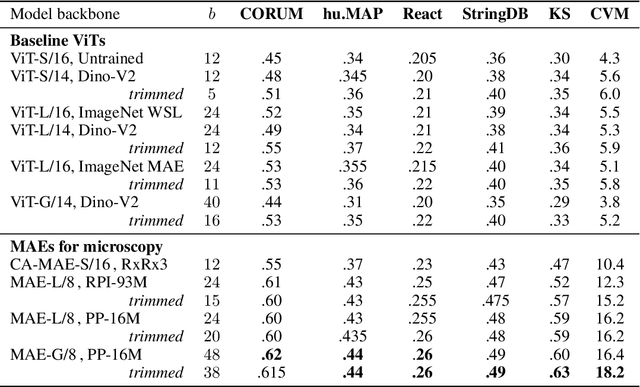
Large-scale cell microscopy screens are used in drug discovery and molecular biology research to study the effects of millions of chemical and genetic perturbations on cells. To use these images in downstream analysis, we need models that can map each image into a feature space that represents diverse biological phenotypes consistently, in the sense that perturbations with similar biological effects have similar representations. In this work, we present the largest foundation model for cell microscopy data to date, a new 1.9 billion-parameter ViT-G/8 MAE trained on over 8 billion microscopy image crops. Compared to a previous published ViT-L/8 MAE, our new model achieves a 60% improvement in linear separability of genetic perturbations and obtains the best overall performance on whole-genome biological relationship recall and replicate consistency benchmarks. Beyond scaling, we developed two key methods that improve performance: (1) training on a curated and diverse dataset; and, (2) using biologically motivated linear probing tasks to search across each transformer block for the best candidate representation of whole-genome screens. We find that many self-supervised vision transformers, pretrained on either natural or microscopy images, yield significantly more biologically meaningful representations of microscopy images in their intermediate blocks than in their typically used final blocks. More broadly, our approach and results provide insights toward a general strategy for successfully building foundation models for large-scale biological data.
Generating counterfactual explanations of tumor spatial proteomes to discover effective, combinatorial therapies that enhance cancer immunotherapy
Nov 08, 2022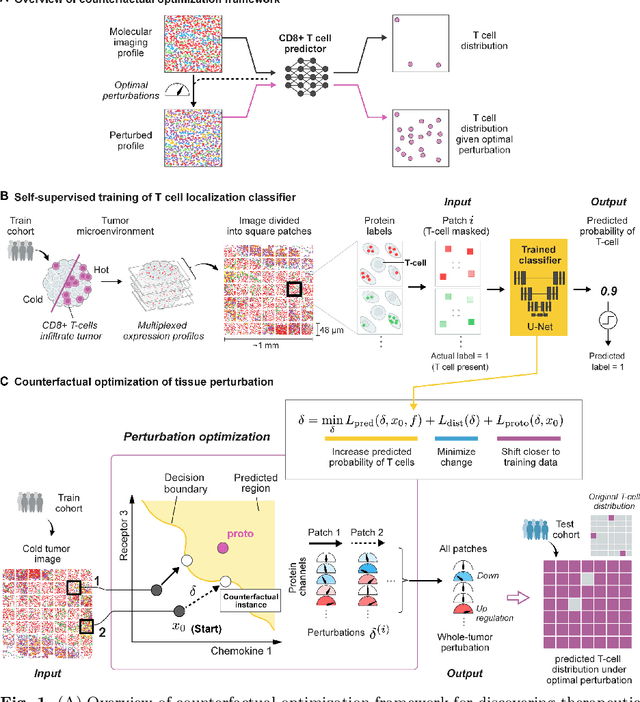

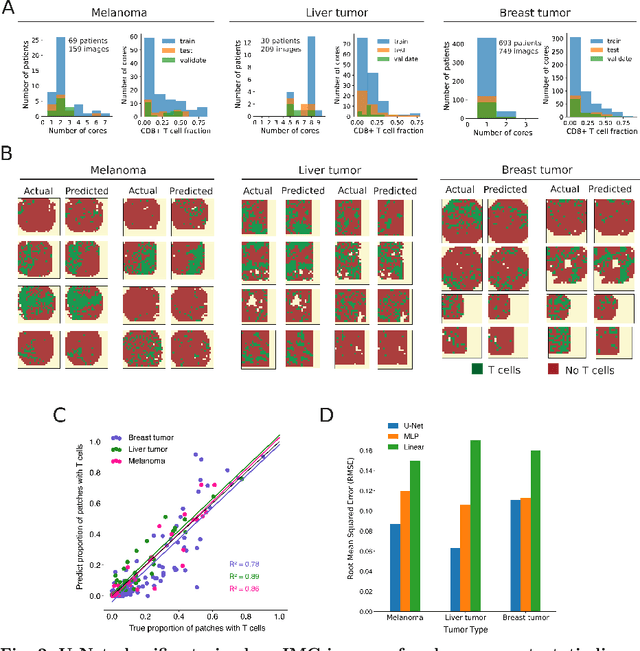

Recent advances in spatial omics methods enable the molecular composition of human tumors to be imaged at micron-scale resolution across hundreds of patients and ten to thousands of molecular imaging channels. Large-scale molecular imaging datasets offer a new opportunity to understand how the spatial organization of proteins and cell types within a tumor modulate the response of a patient to different therapeutic strategies and offer potential insights into the design of novel therapies to increase patient response. However, spatial omics datasets require computational analysis methods that can scale to incorporate hundreds to thousands of imaging channels (ie colors) while enabling the extraction of molecular patterns that correlate with treatment responses across large number of patients with potentially heterogeneous tumors presentations. Here, we have develop a machine learning strategy for the identification and design of signaling molecule combinations that predict the degree of immune system engagement with a specific patient tumors. We specifically train a classifier to predict T cell distribution in patient tumors using the images from 30-40 molecular imaging channels. Second, we apply a gradient descent based counterfactual reasoning strategy to the classifier and discover combinations of signaling molecules predicted to increase T cell infiltration. Applied to spatial proteomics data of melanoma tumor, our model predicts that increasing the level of CXCL9, CXCL10, CXCL12, CCL19 and decreasing the level of CCL8 in melanoma tumor will increase T cell infiltration by 10-fold across a cohort of 69 patients. The model predicts that the combination is many fold more effective than single target perturbations. Our work provides a paradigm for machine learning based prediction and design of cancer therapeutics based on classification of immune system activity in spatial omics data.