 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRxRx3-core: Benchmarking drug-target interactions in High-Content Microscopy
Mar 26, 2025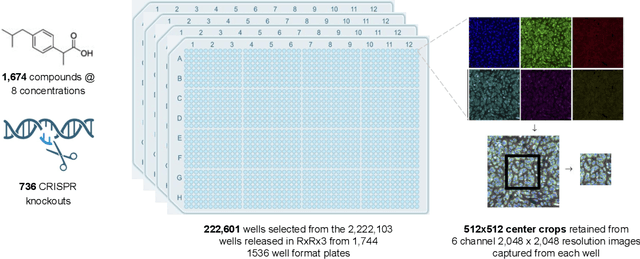


High Content Screening (HCS) microscopy datasets have transformed the ability to profile cellular responses to genetic and chemical perturbations, enabling cell-based inference of drug-target interactions (DTI). However, the adoption of representation learning methods for HCS data has been hindered by the lack of accessible datasets and robust benchmarks. To address this gap, we present RxRx3-core, a curated and compressed subset of the RxRx3 dataset, and an associated DTI benchmarking task. At just 18GB, RxRx3-core significantly reduces the size barrier associated with large-scale HCS datasets while preserving critical data necessary for benchmarking representation learning models against a zero-shot DTI prediction task. RxRx3-core includes 222,601 microscopy images spanning 736 CRISPR knockouts and 1,674 compounds at 8 concentrations. RxRx3-core is available on HuggingFace and Polaris, along with pre-trained embeddings and benchmarking code, ensuring accessibility for the research community. By providing a compact dataset and robust benchmarks, we aim to accelerate innovation in representation learning methods for HCS data and support the discovery of novel biological insights.
ViTally Consistent: Scaling Biological Representation Learning for Cell Microscopy
Nov 04, 2024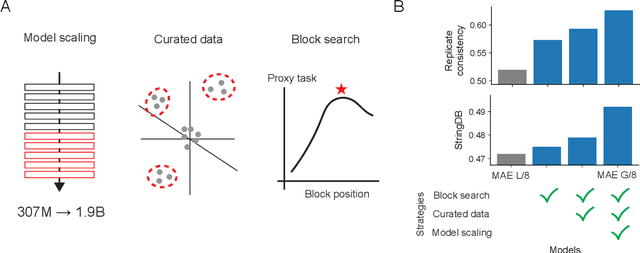
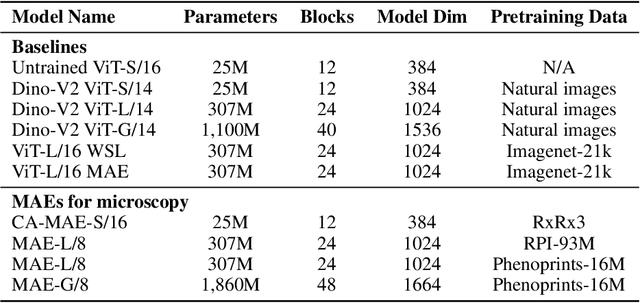
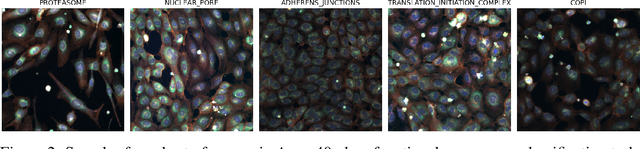
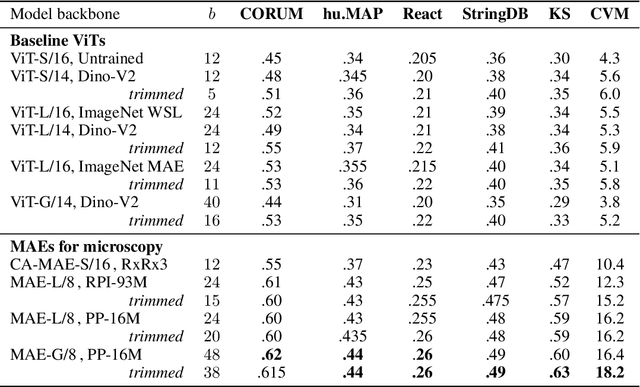
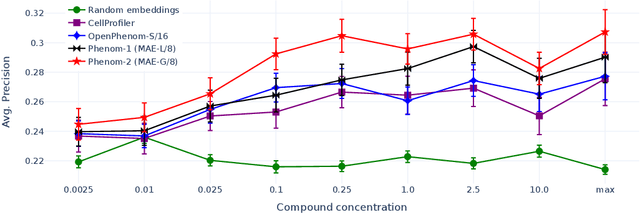
Large-scale cell microscopy screens are used in drug discovery and molecular biology research to study the effects of millions of chemical and genetic perturbations on cells. To use these images in downstream analysis, we need models that can map each image into a feature space that represents diverse biological phenotypes consistently, in the sense that perturbations with similar biological effects have similar representations. In this work, we present the largest foundation model for cell microscopy data to date, a new 1.9 billion-parameter ViT-G/8 MAE trained on over 8 billion microscopy image crops. Compared to a previous published ViT-L/8 MAE, our new model achieves a 60% improvement in linear separability of genetic perturbations and obtains the best overall performance on whole-genome biological relationship recall and replicate consistency benchmarks. Beyond scaling, we developed two key methods that improve performance: (1) training on a curated and diverse dataset; and, (2) using biologically motivated linear probing tasks to search across each transformer block for the best candidate representation of whole-genome screens. We find that many self-supervised vision transformers, pretrained on either natural or microscopy images, yield significantly more biologically meaningful representations of microscopy images in their intermediate blocks than in their typically used final blocks. More broadly, our approach and results provide insights toward a general strategy for successfully building foundation models for large-scale biological data.
RxRx1: A Dataset for Evaluating Experimental Batch Correction Methods
Jan 13, 2023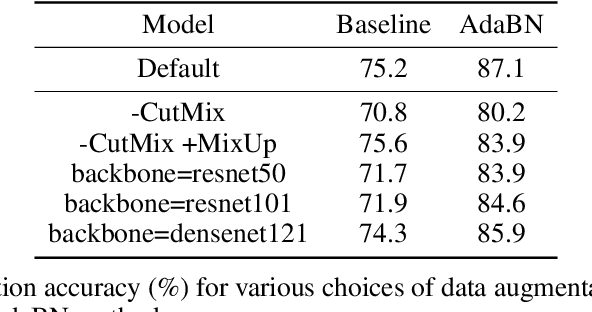



High-throughput screening techniques are commonly used to obtain large quantities of data in many fields of biology. It is well known that artifacts arising from variability in the technical execution of different experimental batches within such screens confound these observations and can lead to invalid biological conclusions. It is therefore necessary to account for these batch effects when analyzing outcomes. In this paper we describe RxRx1, a biological dataset designed specifically for the systematic study of batch effect correction methods. The dataset consists of 125,510 high-resolution fluorescence microscopy images of human cells under 1,138 genetic perturbations in 51 experimental batches across 4 cell types. Visual inspection of the images alone clearly demonstrates significant batch effects. We propose a classification task designed to evaluate the effectiveness of experimental batch correction methods on these images and examine the performance of a number of correction methods on this task. Our goal in releasing RxRx1 is to encourage the development of effective experimental batch correction methods that generalize well to unseen experimental batches. The dataset can be downloaded at https://rxrx.ai.