 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEffective Biological Representation Learning by Masking Gene Expression
May 29, 2026RNA sequencing produces rich and diverse datasets of gene expression, offering compelling insights into cellular state and function that have many applications in drug discovery. Modeling such data is challenging due to inherent technical noise and experimental batch effects, as evidenced by many existing transcriptomic foundation models (FMs) underperforming relative to linear baselines. Such results raise the question of whether deep representation learning provides a distinct advantage over the direct use of raw transcript counts. Our work explores this by developing a new self-supervised model, TxFM, with a focus on inductive representation learning evaluations. TxFM employs a masked autoencoding approach tailored to diverse RNA-seq count data, and our ablation study empirically identifies crucial architecture configurations required for strong transfer performance. Additionally, we curate a public training corpus, DiverseRNA-1.4M, and find that TxFM trained on this curated dataset yields high-fidelity gene representations that outperform FMs trained on atlas-scale corpora over 100x larger. Overall, our results indicate that inductive self-supervised learning is a viable modeling approach for transcriptomics representation, provided a careful synthesis of model architecture and training data curation.
Deep Learning for BioImaging: What Are We Learning?
Mar 10, 2026Representation learning has driven major advances in natural image analysis by enabling models to acquire high-level semantic features. In microscopy imaging, however, it remains unclear what current representation learning methods actually learn. In this work, we conduct a systematic study of representation learning for the two most widely used and broadly available microscopy data types, representing critical scales in biology: cell culture and tissue imaging. To this end, we introduce a set of simple yet revealing baselines on curated benchmarks, including untrained models and simple structural representations of cellular tissue. Our results show that, surprisingly, state-of-the-art methods perform comparably to these baselines. We further show that, in contrast to natural images, existing models fail to consistently acquire high-level, biologically meaningful features. Moreover, we demonstrate that commonly used benchmark metrics are insufficient to assess representation quality and often mask this limitation. In addition, we investigate how detailed comparisons with these benchmarks provide ways to interpret the strengths and weaknesses of models for further improvements. Together, our results suggest that progress in microscopy image representation learning requires not only stronger models, but also more diagnostic benchmarks that measure what is actually learned.
XFACTORS: Disentangled Information Bottleneck via Contrastive Supervision
Jan 29, 2026Disentangled representation learning aims to map independent factors of variation to independent representation components. On one hand, purely unsupervised approaches have proven successful on fully disentangled synthetic data, but fail to recover semantic factors from real data without strong inductive biases. On the other hand, supervised approaches are unstable and hard to scale to large attribute sets because they rely on adversarial objectives or auxiliary classifiers. We introduce \textsc{XFactors}, a weakly-supervised VAE framework that disentangles and provides explicit control over a chosen set of factors. Building on the Disentangled Information Bottleneck perspective, we decompose the representation into a residual subspace $\mathcal{S}$ and factor-specific subspaces $\mathcal{T}_1,\ldots,\mathcal{T}_K$ and a residual subspace $\mathcal{S}$. Each target factor is encoded in its assigned $\mathcal{T}_i$ through contrastive supervision: an InfoNCE loss pulls together latents sharing the same factor value and pushes apart mismatched pairs. In parallel, KL regularization imposes a Gaussian structure on both $\mathcal{S}$ and the aggregated factor subspaces, organizing the geometry without additional supervision for non-targeted factors and avoiding adversarial training and classifiers. Across multiple datasets, with constant hyperparameters, \textsc{XFactors} achieves state-of-the-art disentanglement scores and yields consistent qualitative factor alignment in the corresponding subspaces, enabling controlled factor swapping via latent replacement. We further demonstrate that our method scales correctly with increasing latent capacity and evaluate it on the real-world dataset CelebA. Our code is available at \href{https://github.com/ICML26-anon/XFactors}{github.com/ICML26-anon/XFactors}.
A Cross Modal Knowledge Distillation & Data Augmentation Recipe for Improving Transcriptomics Representations through Morphological Features
May 27, 2025Understanding cellular responses to stimuli is crucial for biological discovery and drug development. Transcriptomics provides interpretable, gene-level insights, while microscopy imaging offers rich predictive features but is harder to interpret. Weakly paired datasets, where samples share biological states, enable multimodal learning but are scarce, limiting their utility for training and multimodal inference. We propose a framework to enhance transcriptomics by distilling knowledge from microscopy images. Using weakly paired data, our method aligns and binds modalities, enriching gene expression representations with morphological information. To address data scarcity, we introduce (1) Semi-Clipped, an adaptation of CLIP for cross-modal distillation using pretrained foundation models, achieving state-of-the-art results, and (2) PEA (Perturbation Embedding Augmentation), a novel augmentation technique that enhances transcriptomics data while preserving inherent biological information. These strategies improve the predictive power and retain the interpretability of transcriptomics, enabling rich unimodal representations for complex biological tasks.
TxPert: Leveraging Biochemical Relationships for Out-of-Distribution Transcriptomic Perturbation Prediction
May 20, 2025Accurately predicting cellular responses to genetic perturbations is essential for understanding disease mechanisms and designing effective therapies. Yet exhaustively exploring the space of possible perturbations (e.g., multi-gene perturbations or across tissues and cell types) is prohibitively expensive, motivating methods that can generalize to unseen conditions. In this work, we explore how knowledge graphs of gene-gene relationships can improve out-of-distribution (OOD) prediction across three challenging settings: unseen single perturbations; unseen double perturbations; and unseen cell lines. In particular, we present: (i) TxPert, a new state-of-the-art method that leverages multiple biological knowledge networks to predict transcriptional responses under OOD scenarios; (ii) an in-depth analysis demonstrating the impact of graphs, model architecture, and data on performance; and (iii) an expanded benchmarking framework that strengthens evaluation standards for perturbation modeling.
ViTally Consistent: Scaling Biological Representation Learning for Cell Microscopy
Nov 04, 2024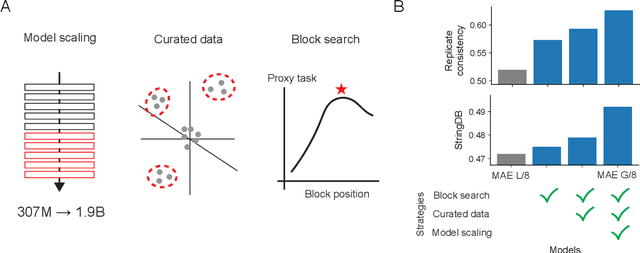
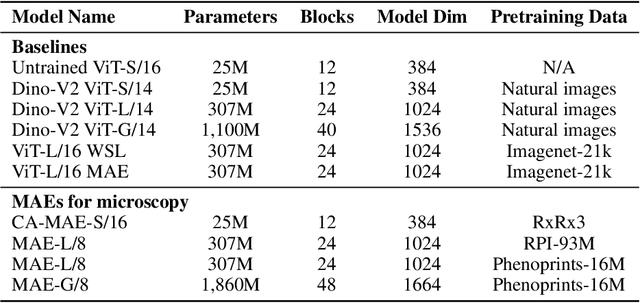
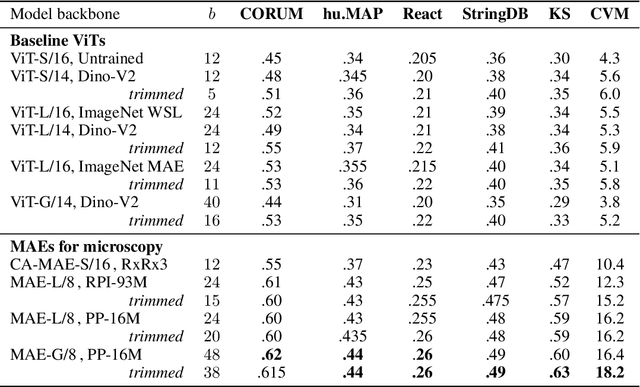
Large-scale cell microscopy screens are used in drug discovery and molecular biology research to study the effects of millions of chemical and genetic perturbations on cells. To use these images in downstream analysis, we need models that can map each image into a feature space that represents diverse biological phenotypes consistently, in the sense that perturbations with similar biological effects have similar representations. In this work, we present the largest foundation model for cell microscopy data to date, a new 1.9 billion-parameter ViT-G/8 MAE trained on over 8 billion microscopy image crops. Compared to a previous published ViT-L/8 MAE, our new model achieves a 60% improvement in linear separability of genetic perturbations and obtains the best overall performance on whole-genome biological relationship recall and replicate consistency benchmarks. Beyond scaling, we developed two key methods that improve performance: (1) training on a curated and diverse dataset; and, (2) using biologically motivated linear probing tasks to search across each transformer block for the best candidate representation of whole-genome screens. We find that many self-supervised vision transformers, pretrained on either natural or microscopy images, yield significantly more biologically meaningful representations of microscopy images in their intermediate blocks than in their typically used final blocks. More broadly, our approach and results provide insights toward a general strategy for successfully building foundation models for large-scale biological data.
Benchmarking Transcriptomics Foundation Models for Perturbation Analysis : one PCA still rules them all
Oct 17, 2024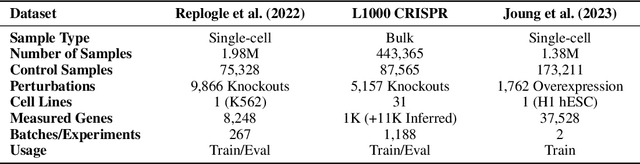
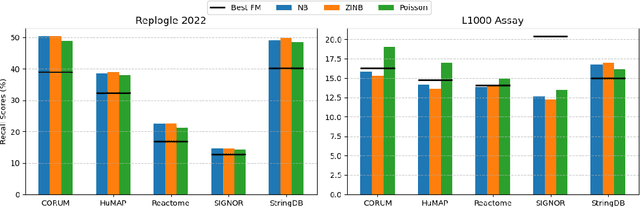
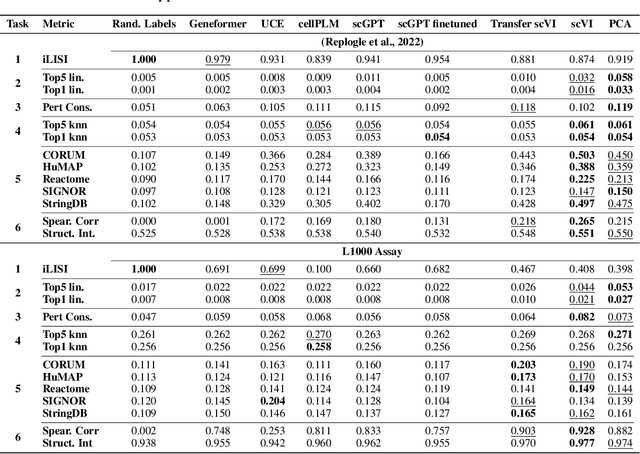
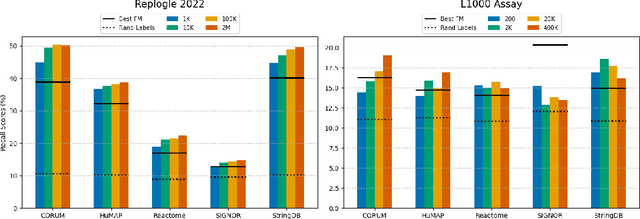
Understanding the relationships among genes, compounds, and their interactions in living organisms remains limited due to technological constraints and the complexity of biological data. Deep learning has shown promise in exploring these relationships using various data types. However, transcriptomics, which provides detailed insights into cellular states, is still underused due to its high noise levels and limited data availability. Recent advancements in transcriptomics sequencing provide new opportunities to uncover valuable insights, especially with the rise of many new foundation models for transcriptomics, yet no benchmark has been made to robustly evaluate the effectiveness of these rising models for perturbation analysis. This article presents a novel biologically motivated evaluation framework and a hierarchy of perturbation analysis tasks for comparing the performance of pretrained foundation models to each other and to more classical techniques of learning from transcriptomics data. We compile diverse public datasets from different sequencing techniques and cell lines to assess models performance. Our approach identifies scVI and PCA to be far better suited models for understanding biological perturbations in comparison to existing foundation models, especially in their application in real-world scenarios.
ChAda-ViT : Channel Adaptive Attention for Joint Representation Learning of Heterogeneous Microscopy Images
Nov 26, 2023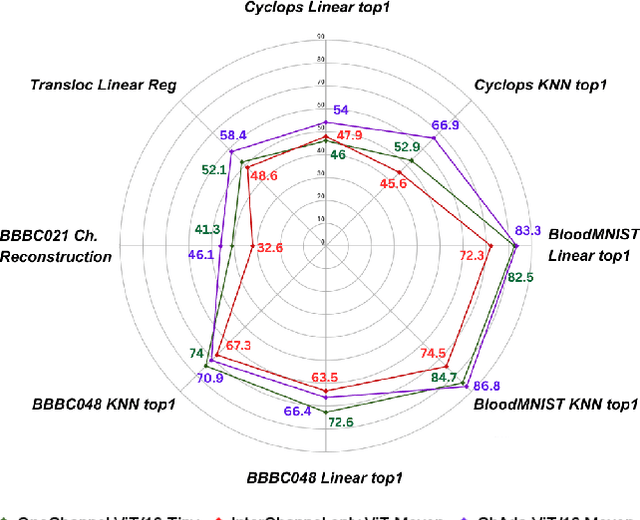
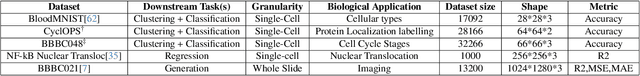
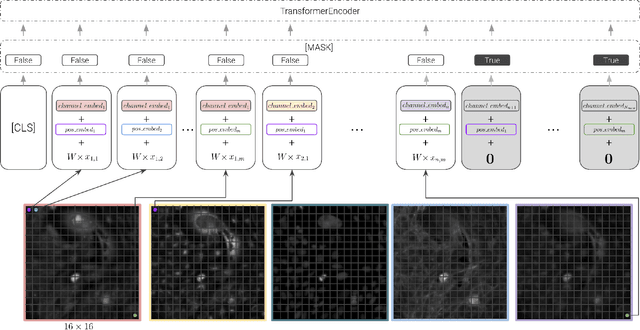

Unlike color photography images, which are consistently encoded into RGB channels, biological images encompass various modalities, where the type of microscopy and the meaning of each channel varies with each experiment. Importantly, the number of channels can range from one to a dozen and their correlation is often comparatively much lower than RGB, as each of them brings specific information content. This aspect is largely overlooked by methods designed out of the bioimage field, and current solutions mostly focus on intra-channel spatial attention, often ignoring the relationship between channels, yet crucial in most biological applications. Importantly, the variable channel type and count prevent the projection of several experiments to a unified representation for large scale pre-training. In this study, we propose ChAda-ViT, a novel Channel Adaptive Vision Transformer architecture employing an Inter-Channel Attention mechanism on images with an arbitrary number, order and type of channels. We also introduce IDRCell100k, a bioimage dataset with a rich set of 79 experiments covering 7 microscope modalities, with a multitude of channel types, and channel counts varying from 1 to 10 per experiment. Our proposed architecture, trained in a self-supervised manner, outperforms existing approaches in several biologically relevant downstream tasks. Additionally, it can be used to bridge the gap for the first time between assays with different microscopes, channel numbers or types by embedding various image and experimental modalities into a unified biological image representation. The latter should facilitate interdisciplinary studies and pave the way for better adoption of deep learning in biological image-based analyses. Code and Data to be released soon.
Prompt Performance Prediction for Generative IR
Jun 15, 2023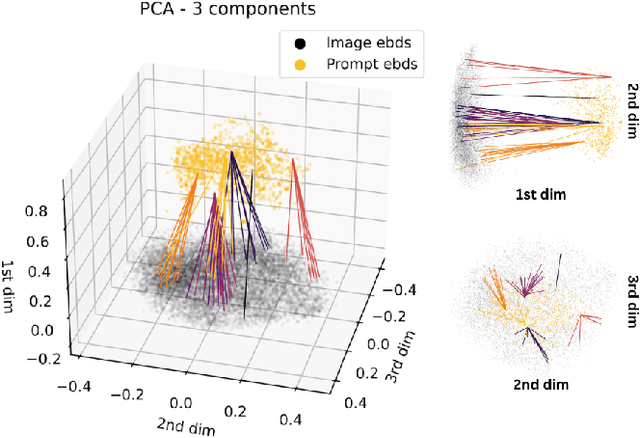

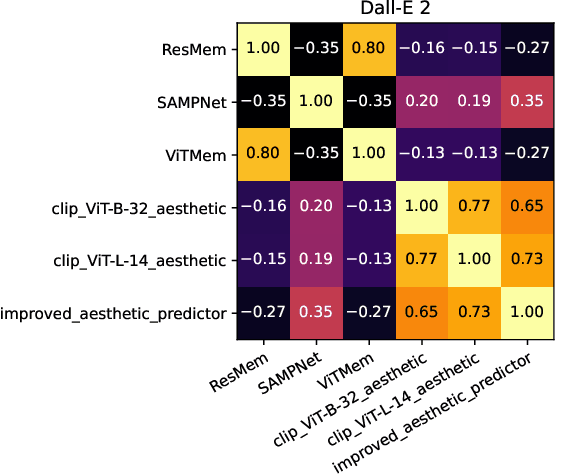
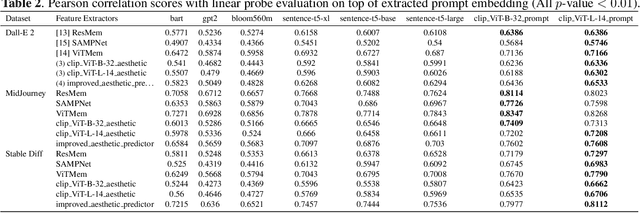
The ability to predict the performance of a query in Information Retrieval (IR) systems has been a longstanding challenge. In this paper, we introduce a novel task called "Prompt Performance Prediction" that aims to predict the performance of a query, referred to as a prompt, before obtaining the actual search results. The context of our task leverages a generative model as an IR engine to evaluate the prompts' performance on image retrieval tasks. We demonstrate the plausibility of our task by measuring the correlation coefficient between predicted and actual performance scores across three datasets containing pairs of prompts and generated images. Our results show promising performance prediction capabilities, suggesting potential applications for optimizing generative IR systems.
No Free Lunch in Self Supervised Representation Learning
Apr 23, 2023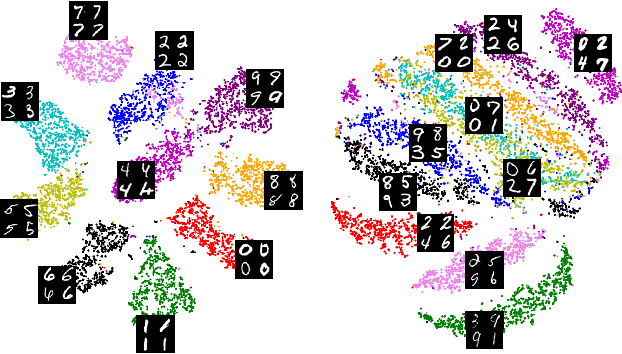

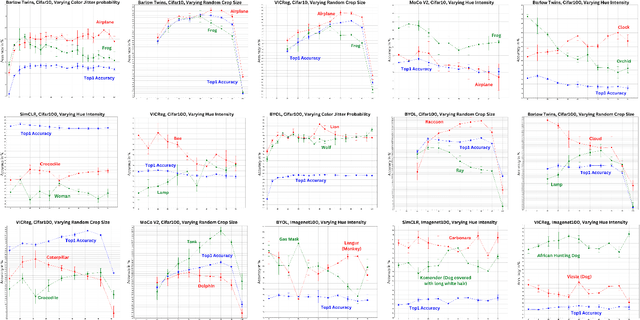
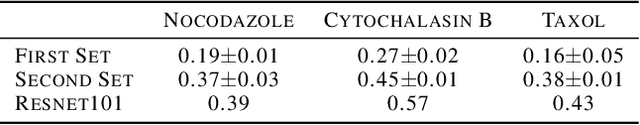
Self-supervised representation learning in computer vision relies heavily on hand-crafted image transformations to learn meaningful and invariant features. However few extensive explorations of the impact of transformation design have been conducted in the literature. In particular, the dependence of downstream performances to transformation design has been established, but not studied in depth. In this work, we explore this relationship, its impact on a domain other than natural images, and show that designing the transformations can be viewed as a form of supervision. First, we demonstrate that not only do transformations have an effect on downstream performance and relevance of clustering, but also that each category in a supervised dataset can be impacted in a different way. Following this, we explore the impact of transformation design on microscopy images, a domain where the difference between classes is more subtle and fuzzy than in natural images. In this case, we observe a greater impact on downstream tasks performances. Finally, we demonstrate that transformation design can be leveraged as a form of supervision, as careful selection of these by a domain expert can lead to a drastic increase in performance on a given downstream task.