 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Cross Modal Knowledge Distillation & Data Augmentation Recipe for Improving Transcriptomics Representations through Morphological Features
May 27, 2025Understanding cellular responses to stimuli is crucial for biological discovery and drug development. Transcriptomics provides interpretable, gene-level insights, while microscopy imaging offers rich predictive features but is harder to interpret. Weakly paired datasets, where samples share biological states, enable multimodal learning but are scarce, limiting their utility for training and multimodal inference. We propose a framework to enhance transcriptomics by distilling knowledge from microscopy images. Using weakly paired data, our method aligns and binds modalities, enriching gene expression representations with morphological information. To address data scarcity, we introduce (1) Semi-Clipped, an adaptation of CLIP for cross-modal distillation using pretrained foundation models, achieving state-of-the-art results, and (2) PEA (Perturbation Embedding Augmentation), a novel augmentation technique that enhances transcriptomics data while preserving inherent biological information. These strategies improve the predictive power and retain the interpretability of transcriptomics, enabling rich unimodal representations for complex biological tasks.
TxPert: Leveraging Biochemical Relationships for Out-of-Distribution Transcriptomic Perturbation Prediction
May 20, 2025Accurately predicting cellular responses to genetic perturbations is essential for understanding disease mechanisms and designing effective therapies. Yet exhaustively exploring the space of possible perturbations (e.g., multi-gene perturbations or across tissues and cell types) is prohibitively expensive, motivating methods that can generalize to unseen conditions. In this work, we explore how knowledge graphs of gene-gene relationships can improve out-of-distribution (OOD) prediction across three challenging settings: unseen single perturbations; unseen double perturbations; and unseen cell lines. In particular, we present: (i) TxPert, a new state-of-the-art method that leverages multiple biological knowledge networks to predict transcriptional responses under OOD scenarios; (ii) an in-depth analysis demonstrating the impact of graphs, model architecture, and data on performance; and (iii) an expanded benchmarking framework that strengthens evaluation standards for perturbation modeling.
SAFE setup for generative molecular design
Oct 26, 2024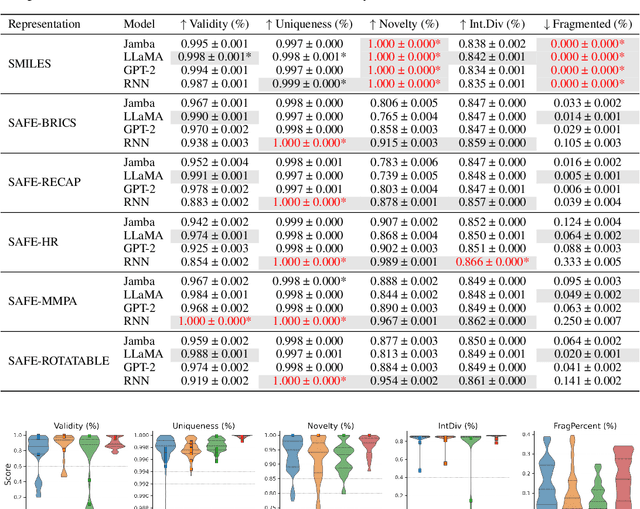
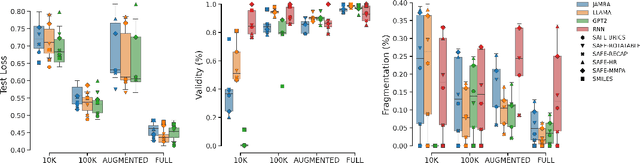
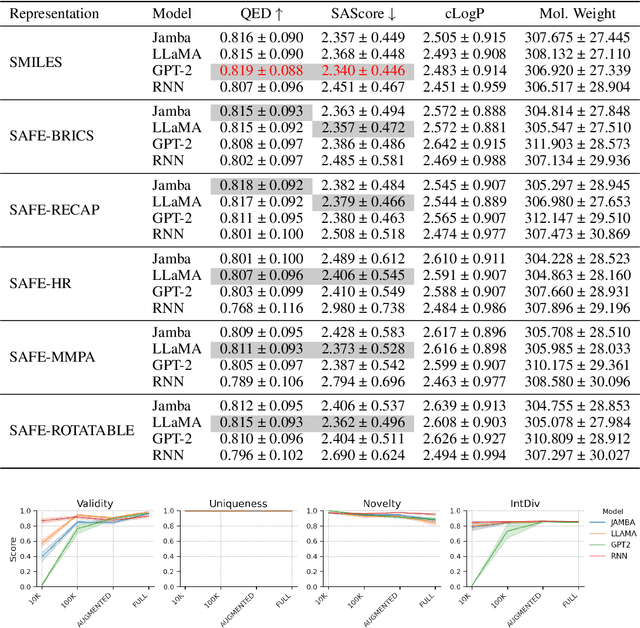
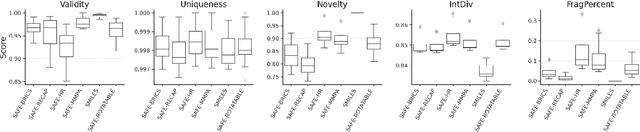
SMILES-based molecular generative models have been pivotal in drug design but face challenges in fragment-constrained tasks. To address this, the Sequential Attachment-based Fragment Embedding (SAFE) representation was recently introduced as an alternative that streamlines those tasks. In this study, we investigate the optimal setups for training SAFE generative models, focusing on dataset size, data augmentation through randomization, model architecture, and bond disconnection algorithms. We found that larger, more diverse datasets improve performance, with the LLaMA architecture using Rotary Positional Embedding proving most robust. SAFE-based models also consistently outperform SMILES-based approaches in scaffold decoration and linker design, particularly with BRICS decomposition yielding the best results. These insights highlight key factors that significantly impact the efficacy of SAFE-based generative models.
Benchmarking Transcriptomics Foundation Models for Perturbation Analysis : one PCA still rules them all
Oct 17, 2024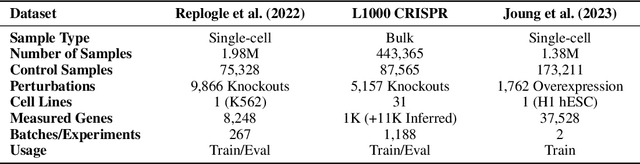
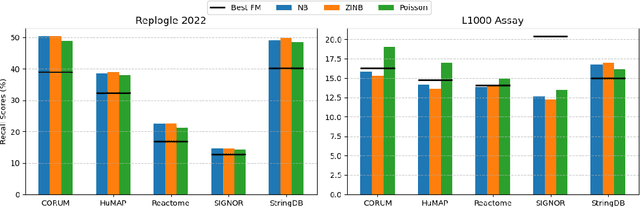
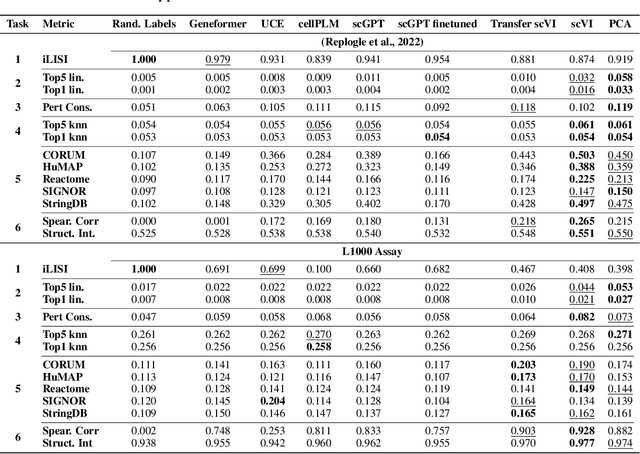
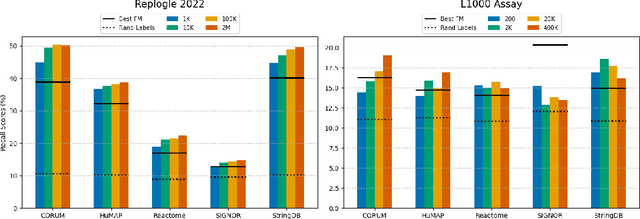
Understanding the relationships among genes, compounds, and their interactions in living organisms remains limited due to technological constraints and the complexity of biological data. Deep learning has shown promise in exploring these relationships using various data types. However, transcriptomics, which provides detailed insights into cellular states, is still underused due to its high noise levels and limited data availability. Recent advancements in transcriptomics sequencing provide new opportunities to uncover valuable insights, especially with the rise of many new foundation models for transcriptomics, yet no benchmark has been made to robustly evaluate the effectiveness of these rising models for perturbation analysis. This article presents a novel biologically motivated evaluation framework and a hierarchy of perturbation analysis tasks for comparing the performance of pretrained foundation models to each other and to more classical techniques of learning from transcriptomics data. We compile diverse public datasets from different sequencing techniques and cell lines to assess models performance. Our approach identifies scVI and PCA to be far better suited models for understanding biological perturbations in comparison to existing foundation models, especially in their application in real-world scenarios.
A Short Study on Compressing Decoder-Based Language Models
Oct 16, 2021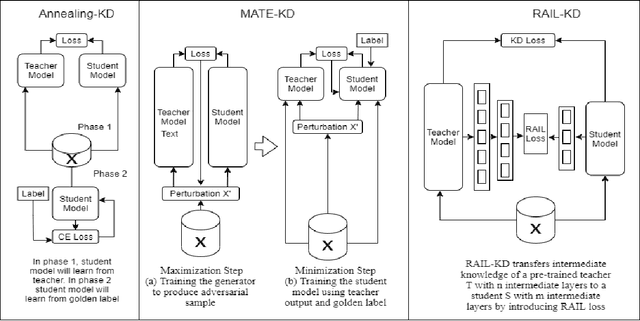
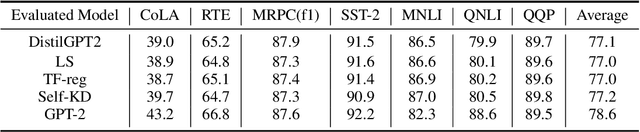
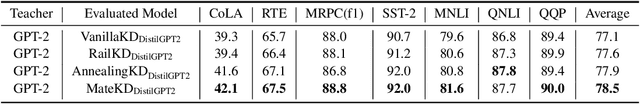
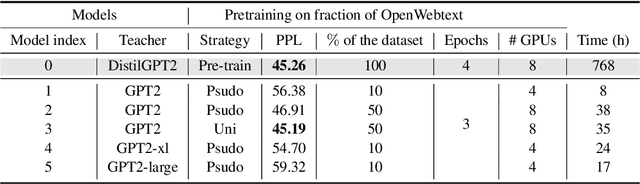
Pre-trained Language Models (PLMs) have been successful for a wide range of natural language processing (NLP) tasks. The state-of-the-art of PLMs, however, are extremely large to be used on edge devices. As a result, the topic of model compression has attracted increasing attention in the NLP community. Most of the existing works focus on compressing encoder-based models (tiny-BERT, distilBERT, distilRoBERTa, etc), however, to the best of our knowledge, the compression of decoder-based models (such as GPT-2) has not been investigated much. Our paper aims to fill this gap. Specifically, we explore two directions: 1) we employ current state-of-the-art knowledge distillation techniques to improve fine-tuning of DistilGPT-2. 2) we pre-train a compressed GPT-2 model using layer truncation and compare it against the distillation-based method (DistilGPT2). The training time of our compressed model is significantly less than DistilGPT-2, but it can achieve better performance when fine-tuned on downstream tasks. We also demonstrate the impact of data cleaning on model performance.