 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking Transcriptomics Foundation Models for Perturbation Analysis : one PCA still rules them all
Paper and Code
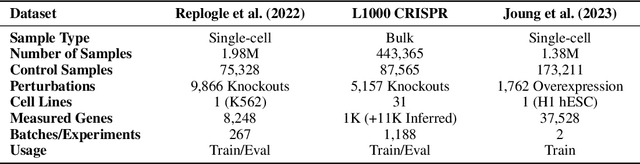
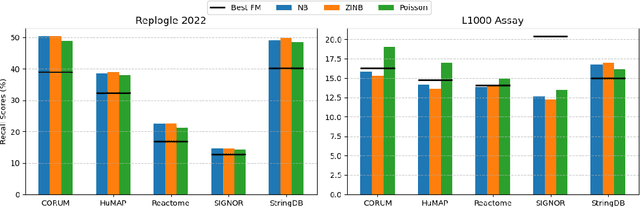
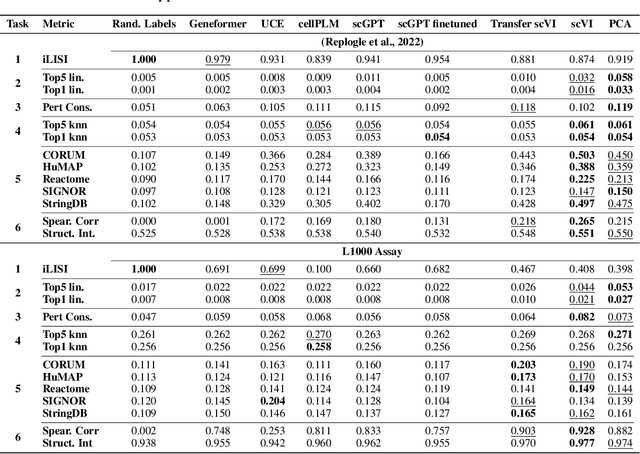
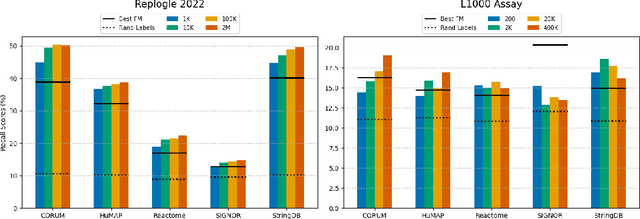
Understanding the relationships among genes, compounds, and their interactions in living organisms remains limited due to technological constraints and the complexity of biological data. Deep learning has shown promise in exploring these relationships using various data types. However, transcriptomics, which provides detailed insights into cellular states, is still underused due to its high noise levels and limited data availability. Recent advancements in transcriptomics sequencing provide new opportunities to uncover valuable insights, especially with the rise of many new foundation models for transcriptomics, yet no benchmark has been made to robustly evaluate the effectiveness of these rising models for perturbation analysis. This article presents a novel biologically motivated evaluation framework and a hierarchy of perturbation analysis tasks for comparing the performance of pretrained foundation models to each other and to more classical techniques of learning from transcriptomics data. We compile diverse public datasets from different sequencing techniques and cell lines to assess models performance. Our approach identifies scVI and PCA to be far better suited models for understanding biological perturbations in comparison to existing foundation models, especially in their application in real-world scenarios.