 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioResearcher: Scenario-Guided Multi-Agent for Translational Medicine
May 07, 2026Translational medicine turns underspecified development goals into evidence synthesis that must combine literature, trials, patents, and quantitative multi-omics analysis while preserving identifiers, uncertainty, and retrievable provenance. General-purpose foundation models and off-the-shelf tool-augmented or multi-agent systems are not built for this: they tend to produce single-shot answers or run open-endedly, and fall short on the auditable, scenario-specific workflows that heterogeneous biomedical sources demand. This paper introduces Ingenix BioResearcher, a scenario-guided multi-agent system that maps queries to versioned research playbooks, delegates to specialized subagents over 30+ tools and machine-learning endpoints, mixes structured database access with sandboxed code for genome-scale analyses, and applies claim-level multi-model reconciliation before editorial assembly. We evaluate BioResearcher across unit-level capabilities, open-ended biomedical reasoning, and end-to-end clinical discovery. It leads evaluated baselines on 109 single-step tests (83.49% pass rate; 0.892 average score), achieves strong biomedical benchmark performance (89.33% on BixBench-Verified-50 and the top 0.758 mean score on BaisBench Scientific Discovery), and leads on a 30-query clinical end-to-end benchmark with the highest positive hit rate (74.7% $\pm$ 3.3%) and negative clear rate (96.8% $\pm$ 0.2%). These results show broad, competitive performance across unit-level, open-ended, and end-to-end clinical evaluations.
On the Scalability of GNNs for Molecular Graphs
Apr 17, 2024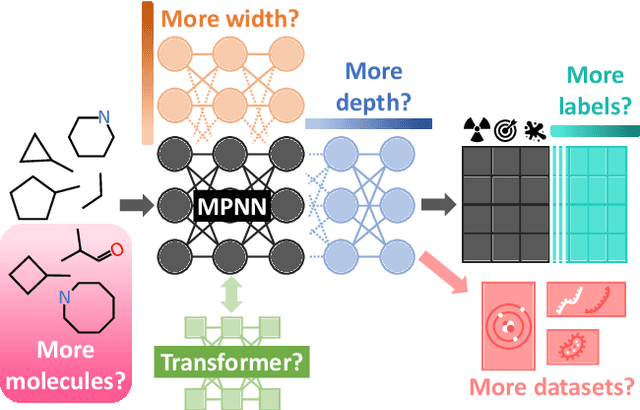

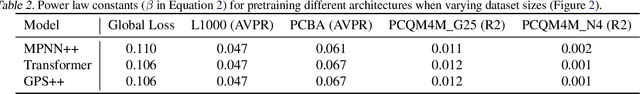
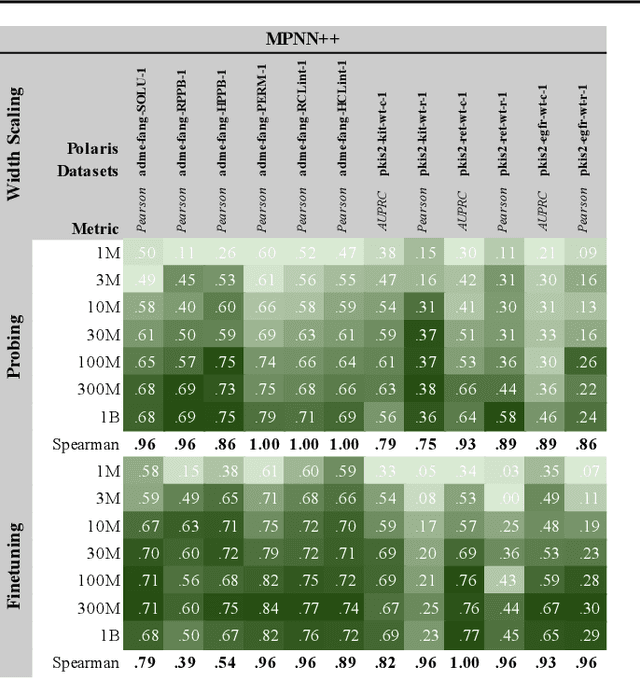
Scaling deep learning models has been at the heart of recent revolutions in language modelling and image generation. Practitioners have observed a strong relationship between model size, dataset size, and performance. However, structure-based architectures such as Graph Neural Networks (GNNs) are yet to show the benefits of scale mainly due to the lower efficiency of sparse operations, large data requirements, and lack of clarity about the effectiveness of various architectures. We address this drawback of GNNs by studying their scaling behavior. Specifically, we analyze message-passing networks, graph Transformers, and hybrid architectures on the largest public collection of 2D molecular graphs. For the first time, we observe that GNNs benefit tremendously from the increasing scale of depth, width, number of molecules, number of labels, and the diversity in the pretraining datasets, resulting in a 30.25% improvement when scaling to 1 billion parameters and 28.98% improvement when increasing size of dataset to eightfold. We further demonstrate strong finetuning scaling behavior on 38 tasks, outclassing previous large models. We hope that our work paves the way for an era where foundational GNNs drive pharmaceutical drug discovery.
Masked Autoencoders for Microscopy are Scalable Learners of Cellular Biology
Apr 16, 2024



Featurizing microscopy images for use in biological research remains a significant challenge, especially for large-scale experiments spanning millions of images. This work explores the scaling properties of weakly supervised classifiers and self-supervised masked autoencoders (MAEs) when training with increasingly larger model backbones and microscopy datasets. Our results show that ViT-based MAEs outperform weakly supervised classifiers on a variety of tasks, achieving as much as a 11.5% relative improvement when recalling known biological relationships curated from public databases. Additionally, we develop a new channel-agnostic MAE architecture (CA-MAE) that allows for inputting images of different numbers and orders of channels at inference time. We demonstrate that CA-MAEs effectively generalize by inferring and evaluating on a microscopy image dataset (JUMP-CP) generated under different experimental conditions with a different channel structure than our pretraining data (RPI-93M). Our findings motivate continued research into scaling self-supervised learning on microscopy data in order to create powerful foundation models of cellular biology that have the potential to catalyze advancements in drug discovery and beyond.
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
Masked autoencoders are scalable learners of cellular morphology
Sep 27, 2023
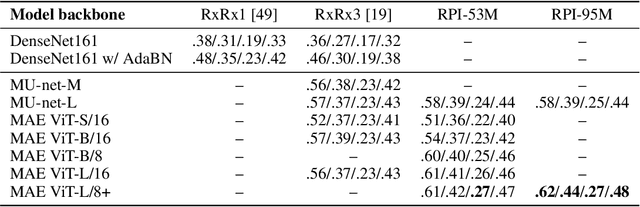

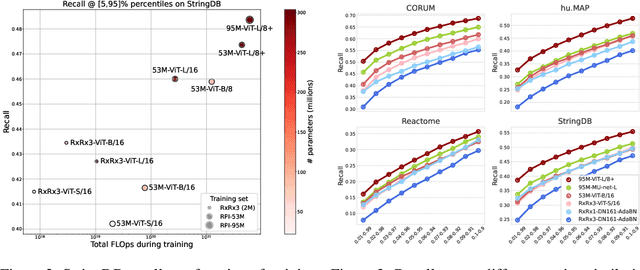
Inferring biological relationships from cellular phenotypes in high-content microscopy screens provides significant opportunity and challenge in biological research. Prior results have shown that deep vision models can capture biological signal better than hand-crafted features. This work explores how weakly supervised and self-supervised deep learning approaches scale when training larger models on larger datasets. Our results show that both CNN- and ViT-based masked autoencoders significantly outperform weakly supervised models. At the high-end of our scale, a ViT-L/8 trained on over 3.5-billion unique crops sampled from 95-million microscopy images achieves relative improvements as high as 28% over our best weakly supervised models at inferring known biological relationships curated from public databases.
Spatial Latent Representations in Generative Adversarial Networks for Image Generation
Mar 25, 2023In the majority of GAN architectures, the latent space is defined as a set of vectors of given dimensionality. Such representations are not easily interpretable and do not capture spatial information of image content directly. In this work, we define a family of spatial latent spaces for StyleGAN2, capable of capturing more details and representing images that are out-of-sample in terms of the number and arrangement of object parts, such as an image of multiple faces or a face with more than two eyes. We propose a method for encoding images into our spaces, together with an attribute model capable of performing attribute editing in these spaces. We show that our spaces are effective for image manipulation and encode semantic information well. Our approach can be used on pre-trained generator models, and attribute edition can be done using pre-generated direction vectors making the barrier to entry for experimentation and use extremely low. We propose a regularization method for optimizing latent representations, which equalizes distributions of parts of latent spaces, making representations much closer to generated ones. We use it for encoding images into spatial spaces to obtain significant improvement in quality while keeping semantics and ability to use our attribute model for edition purposes. In total, using our methods gives encoding quality boost even as high as 30% in terms of LPIPS score comparing to standard methods, while keeping semantics. Additionally, we propose a StyleGAN2 training procedure on our spatial latent spaces, together with a custom spatial latent representation distribution to make spatially closer elements in the representation more dependent on each other than farther elements. Such approach improves the FID score by 29% on SpaceNet, and is able to generate consistent images of arbitrary sizes on spatially homogeneous datasets, like satellite imagery.
RxRx1: A Dataset for Evaluating Experimental Batch Correction Methods
Jan 13, 2023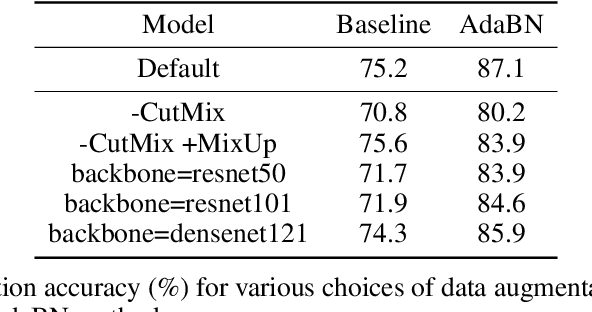



High-throughput screening techniques are commonly used to obtain large quantities of data in many fields of biology. It is well known that artifacts arising from variability in the technical execution of different experimental batches within such screens confound these observations and can lead to invalid biological conclusions. It is therefore necessary to account for these batch effects when analyzing outcomes. In this paper we describe RxRx1, a biological dataset designed specifically for the systematic study of batch effect correction methods. The dataset consists of 125,510 high-resolution fluorescence microscopy images of human cells under 1,138 genetic perturbations in 51 experimental batches across 4 cell types. Visual inspection of the images alone clearly demonstrates significant batch effects. We propose a classification task designed to evaluate the effectiveness of experimental batch correction methods on these images and examine the performance of a number of correction methods on this task. Our goal in releasing RxRx1 is to encourage the development of effective experimental batch correction methods that generalize well to unseen experimental batches. The dataset can be downloaded at https://rxrx.ai.
Insights From the NeurIPS 2021 NetHack Challenge
Mar 22, 2022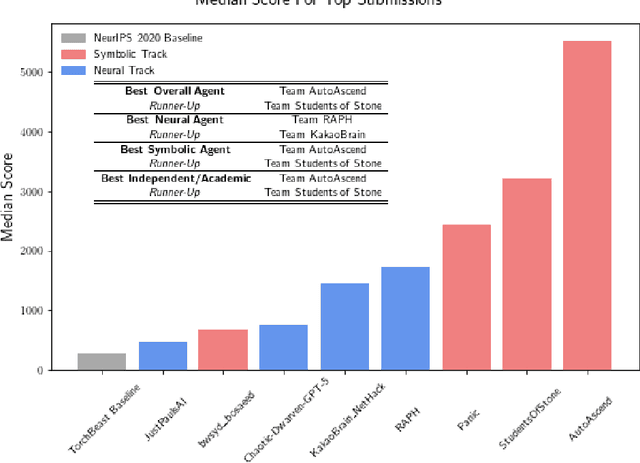
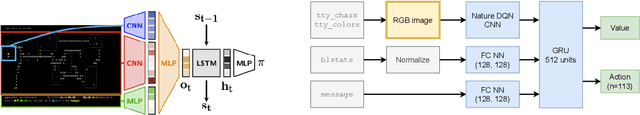
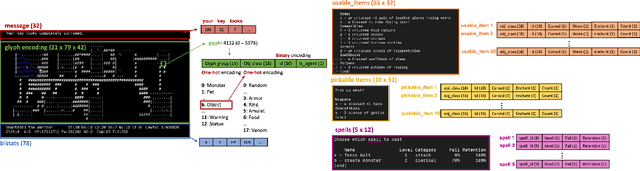
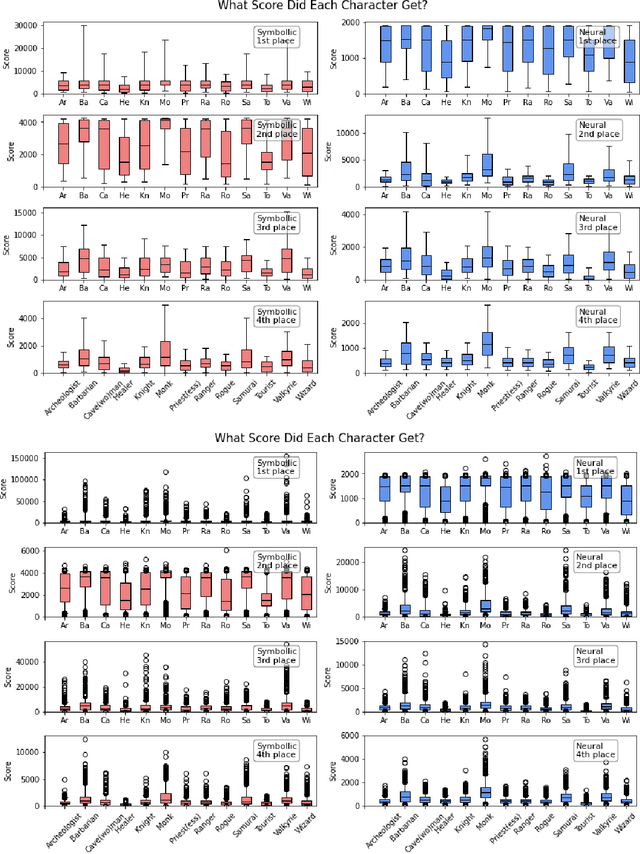
In this report, we summarize the takeaways from the first NeurIPS 2021 NetHack Challenge. Participants were tasked with developing a program or agent that can win (i.e., 'ascend' in) the popular dungeon-crawler game of NetHack by interacting with the NetHack Learning Environment (NLE), a scalable, procedurally generated, and challenging Gym environment for reinforcement learning (RL). The challenge showcased community-driven progress in AI with many diverse approaches significantly beating the previously best results on NetHack. Furthermore, it served as a direct comparison between neural (e.g., deep RL) and symbolic AI, as well as hybrid systems, demonstrating that on NetHack symbolic bots currently outperform deep RL by a large margin. Lastly, no agent got close to winning the game, illustrating NetHack's suitability as a long-term benchmark for AI research.
Augmentation Inside the Network
Dec 19, 2020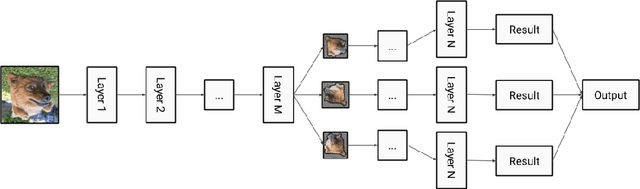
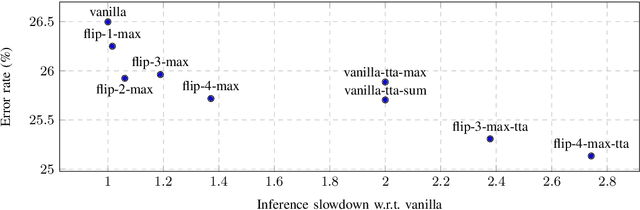
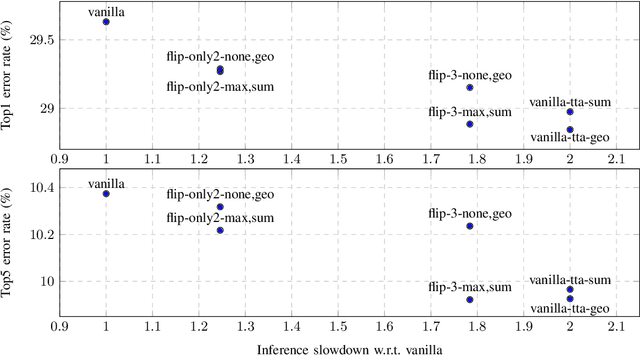
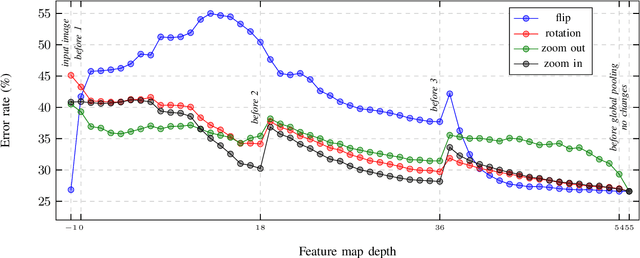
In this paper, we present augmentation inside the network, a method that simulates data augmentation techniques for computer vision problems on intermediate features of a convolutional neural network. We perform these transformations, changing the data flow through the network, and sharing common computations when it is possible. Our method allows us to obtain smoother speed-accuracy trade-off adjustment and achieves better results than using standard test-time augmentation (TTA) techniques. Additionally, our approach can improve model performance even further when coupled with test-time augmentation. We validate our method on the ImageNet-2012 and CIFAR-100 datasets for image classification. We propose a modification that is 30% faster than the flip test-time augmentation and achieves the same results for CIFAR-100.