 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
Gotta be SAFE: A New Framework for Molecular Design
Oct 16, 2023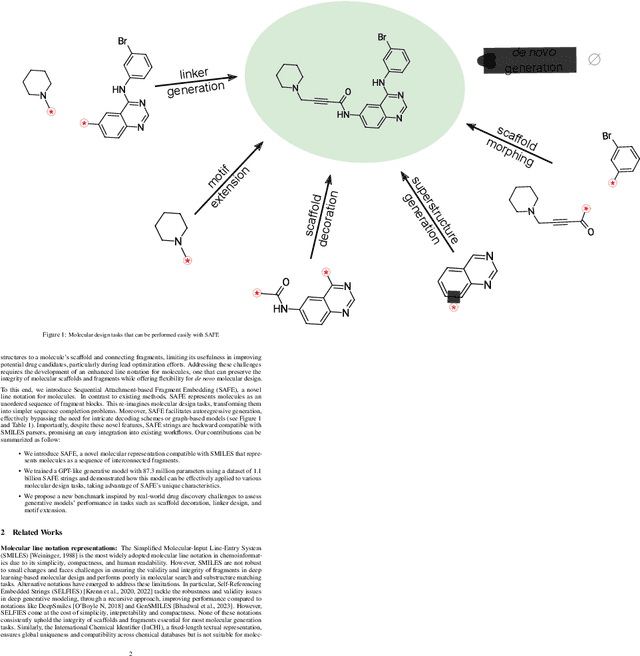

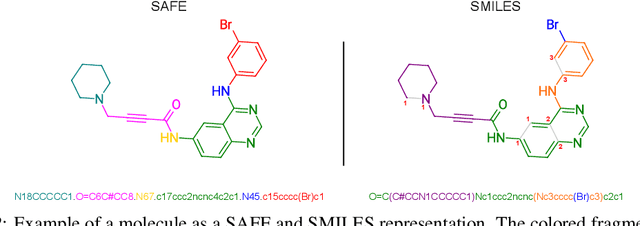
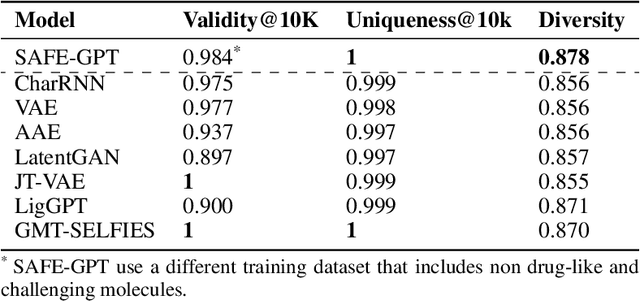
Traditional molecular string representations, such as SMILES, often pose challenges for AI-driven molecular design due to their non-sequential depiction of molecular substructures. To address this issue, we introduce Sequential Attachment-based Fragment Embedding (SAFE), a novel line notation for chemical structures. SAFE reimagines SMILES strings as an unordered sequence of interconnected fragment blocks while maintaining full compatibility with existing SMILES parsers. It streamlines complex generative tasks, including scaffold decoration, fragment linking, polymer generation, and scaffold hopping, while facilitating autoregressive generation for fragment-constrained design, thereby eliminating the need for intricate decoding or graph-based models. We demonstrate the effectiveness of SAFE by training an 87-million-parameter GPT2-like model on a dataset containing 1.1 billion SAFE representations. Through extensive experimentation, we show that our SAFE-GPT model exhibits versatile and robust optimization performance. SAFE opens up new avenues for the rapid exploration of chemical space under various constraints, promising breakthroughs in AI-driven molecular design.
Reconstruction of Long-Term Historical Demand Data
Sep 10, 2022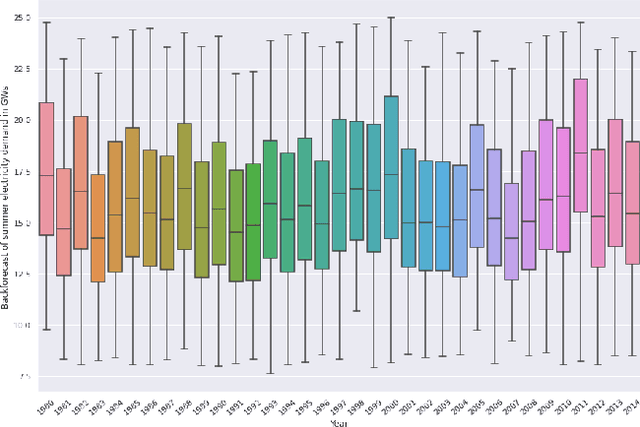

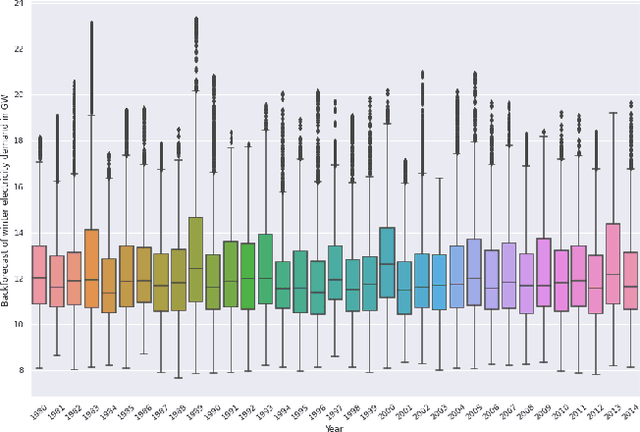
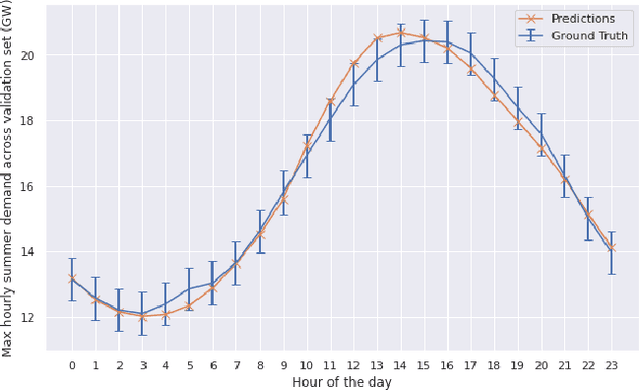
Long-term planning of a robust power system requires the understanding of changing demand patterns. Electricity demand is highly weather sensitive. Thus, the supply side variation from introducing intermittent renewable sources, juxtaposed with variable demand, will introduce additional challenges in the grid planning process. By understanding the spatial and temporal variability of temperature over the US, the response of demand to natural variability and climate change-related effects on temperature can be separated, especially because the effects due to the former factor are not known. Through this project, we aim to better support the technology & policy development process for power systems by developing machine and deep learning 'back-forecasting' models to reconstruct multidecadal demand records and study the natural variability of temperature and its influence on demand.
* Accepted to Tackling Climate Change with Machine Learning Workshop, ICML 2021
Sparse hierarchical representation learning on molecular graphs
Aug 06, 2019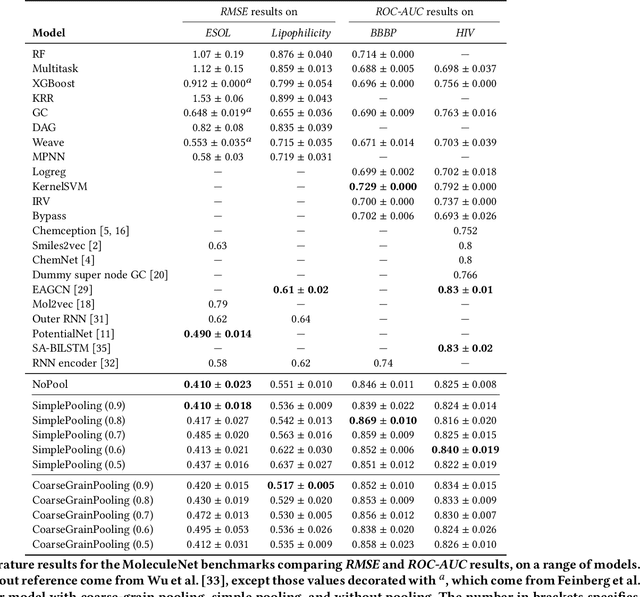
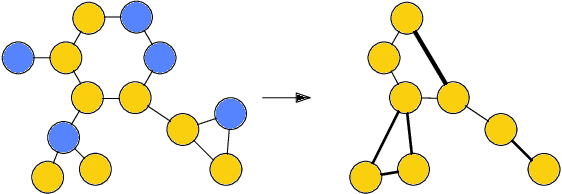

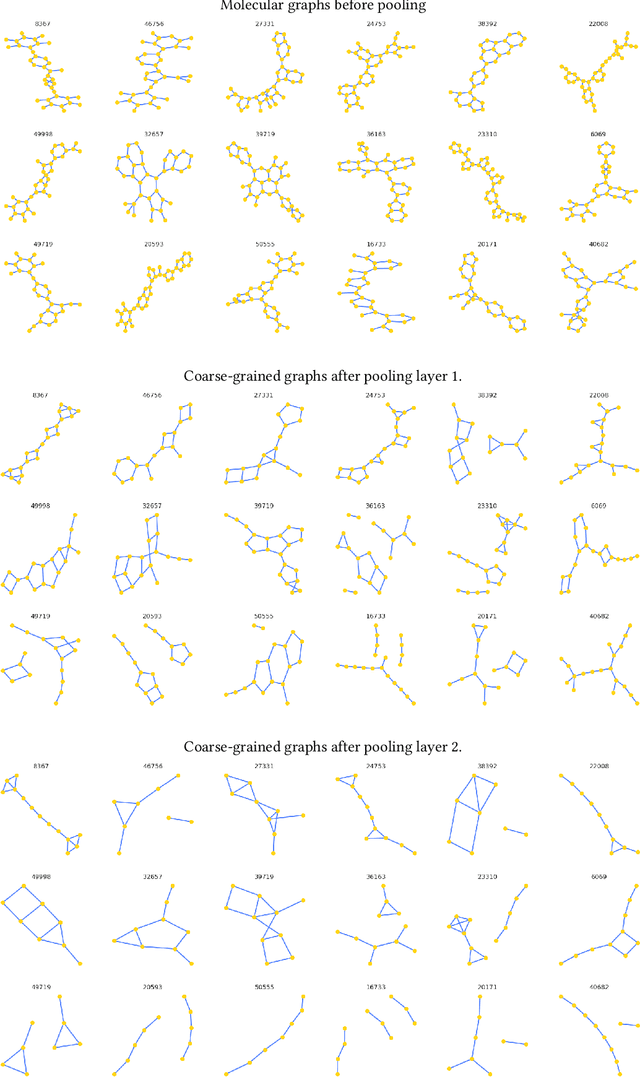
Architectures for sparse hierarchical representation learning have recently been proposed for graph-structured data, but so far assume the absence of edge features in the graph. We close this gap and propose a method to pool graphs with edge features, inspired by the hierarchical nature of chemistry. In particular, we introduce two types of pooling layers compatible with an edge-feature graph-convolutional architecture and investigate their performance for molecules relevant to drug discovery on a set of two classification and two regression benchmark datasets of MoleculeNet. We find that our models significantly outperform previous benchmarks on three of the datasets and reach state-of-the-art results on the fourth benchmark, with pooling improving performance for three out of four tasks, keeping performance stable on the fourth task, and generally speeding up the training process.