 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStochastic Interpolants in Hilbert Spaces
Feb 02, 2026Although diffusion models have successfully extended to function-valued data, stochastic interpolants -- which offer a flexible way to bridge arbitrary distributions -- remain limited to finite-dimensional settings. This work bridges this gap by establishing a rigorous framework for stochastic interpolants in infinite-dimensional Hilbert spaces. We provide comprehensive theoretical foundations, including proofs of well-posedness and explicit error bounds. We demonstrate the effectiveness of the proposed framework for conditional generation, focusing particularly on complex PDE-based benchmarks. By enabling generative bridges between arbitrary functional distributions, our approach achieves state-of-the-art results, offering a powerful, general-purpose tool for scientific discovery.
Feature Attribution with Necessity and Sufficiency via Dual-stage Perturbation Test for Causal Explanation
Feb 13, 2024We investigate the problem of explainability in machine learning.To address this problem, Feature Attribution Methods (FAMs) measure the contribution of each feature through a perturbation test, where the difference in prediction is compared under different perturbations.However, such perturbation tests may not accurately distinguish the contributions of different features, when their change in prediction is the same after perturbation.In order to enhance the ability of FAMs to distinguish different features' contributions in this challenging setting, we propose to utilize the probability (PNS) that perturbing a feature is a necessary and sufficient cause for the prediction to change as a measure of feature importance.Our approach, Feature Attribution with Necessity and Sufficiency (FANS), computes the PNS via a perturbation test involving two stages (factual and interventional).In practice, to generate counterfactual samples, we use a resampling-based approach on the observed samples to approximate the required conditional distribution.Finally, we combine FANS and gradient-based optimization to extract the subset with the largest PNS.We demonstrate that FANS outperforms existing feature attribution methods on six benchmarks.
Leveraging Task Structures for Improved Identifiability in Neural Network Representations
Jun 26, 2023This work extends the theory of identifiability in supervised learning by considering the consequences of having access to a distribution of tasks. In such cases, we show that identifiability is achievable even in the case of regression, extending prior work restricted to the single-task classification case. Furthermore, we show that the existence of a task distribution which defines a conditional prior over latent variables reduces the equivalence class for identifiability to permutations and scaling, a much stronger and more useful result. When we further assume a causal structure over these tasks, our approach enables simple maximum marginal likelihood optimization together with downstream applicability to causal representation learning. Empirically, we validate that our model outperforms more general unsupervised models in recovering canonical representations for synthetic and real-world data.
Molecular Design in Synthetically Accessible Chemical Space via Deep Reinforcement Learning
Apr 29, 2020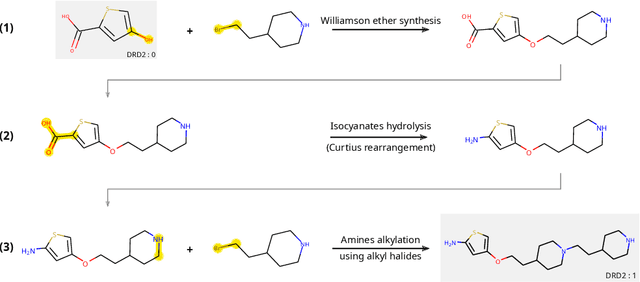
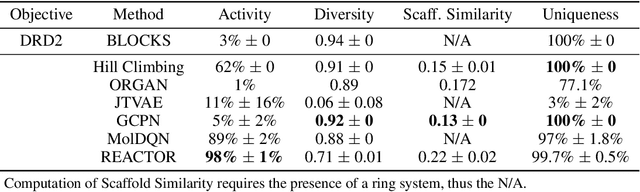
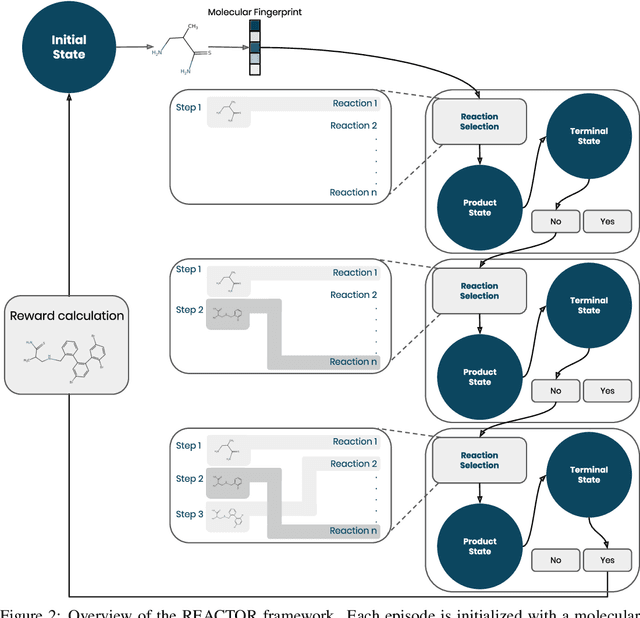
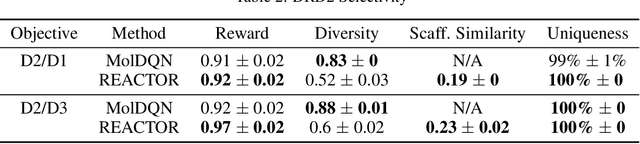
The fundamental goal of generative drug design is to propose optimized molecules that meet predefined activity, selectivity, and pharmacokinetic criteria. Despite recent progress, we argue that existing generative methods are limited in their ability to favourably shift the distributions of molecular properties during optimization. We instead propose a novel Reinforcement Learning framework for molecular design in which an agent learns to directly optimize through a space of synthetically-accessible drug-like molecules. This becomes possible by defining transitions in our Markov Decision Process as chemical reactions, and allows us to leverage synthetic routes as an inductive bias. We validate our method by demonstrating that it outperforms existing state-of the art approaches in the optimization of pharmacologically-relevant objectives, while results on multi-objective optimization tasks suggest increased scalability to realistic pharmaceutical design problems.
Towards Interpretable Sparse Graph Representation Learning with Laplacian Pooling
May 28, 2019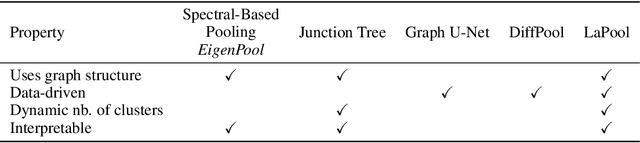
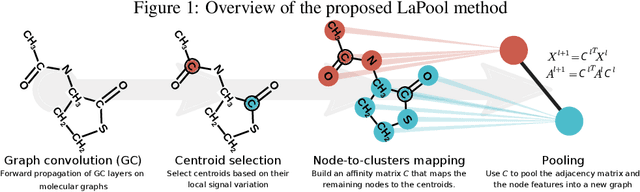
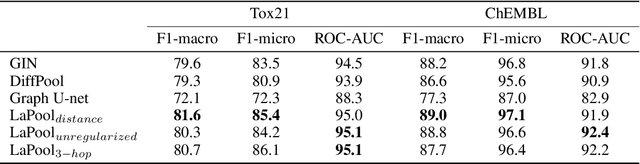
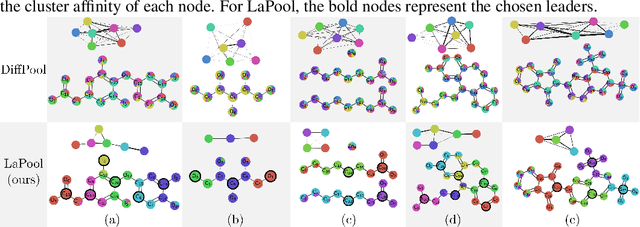
Recent work in graph neural networks (GNNs) has lead to improvements in molecular activity and property prediction tasks. However, GNNs lack interpretability as they fail to capture the relative importance of various molecular substructures due to the absence of efficient intermediate pooling steps for sparse graphs. To address this issue, we propose LaPool (Laplacian Pooling), a novel, data-driven, and interpretable graph pooling method that takes into account the node features and graph structure to improve molecular understanding. Inspired by theories in graph signal processing, LaPool performs a feature-driven hierarchical segmentation of molecules by selecting a set of centroid nodes from a graph as cluster representatives. It then learns a sparse assignment of remaining nodes into these clusters using an attention mechanism. We benchmark our model by showing that it outperforms recent graph pooling layers on molecular graph understanding and prediction tasks. We then demonstrate improved interpretability by identifying important molecular substructures and generating novel and valid molecules, with important applications in drug discovery and pharmacology.