 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultitask finetuning and acceleration of chemical pretrained models for small molecule drug property prediction
Oct 14, 2025Chemical pretrained models, sometimes referred to as foundation models, are receiving considerable interest for drug discovery applications. The general chemical knowledge extracted from self-supervised training has the potential to improve predictions for critical drug discovery endpoints, including on-target potency and ADMET properties. Multi-task learning has previously been successfully leveraged to improve predictive models. Here, we show that enabling multitasking in finetuning of chemical pretrained graph neural network models such as Kinetic GROVER Multi-Task (KERMT), an enhanced version of the GROVER model, and Knowledge-guided Pre-training of Graph Transformer (KGPT) significantly improves performance over non-pretrained graph neural network models. Surprisingly, we find that the performance improvement from finetuning KERMT in a multitask manner is most significant at larger data sizes. Additionally, we publish two multitask ADMET data splits to enable more accurate benchmarking of multitask deep learning methods for drug property prediction. Finally, we provide an accelerated implementation of the KERMT model on GitHub, unlocking large-scale pretraining, finetuning, and inference in industrial drug discovery workflows.
Rethinking Molecule Synthesizability with Chain-of-Reaction
Sep 19, 2025A well-known pitfall of molecular generative models is that they are not guaranteed to generate synthesizable molecules. There have been considerable attempts to address this problem, but given the exponentially large combinatorial space of synthesizable molecules, existing methods have shown limited coverage of the space and poor molecular optimization performance. To tackle these problems, we introduce ReaSyn, a generative framework for synthesizable projection where the model explores the neighborhood of given molecules in the synthesizable space by generating pathways that result in synthesizable analogs. To fully utilize the chemical knowledge contained in the synthetic pathways, we propose a novel perspective that views synthetic pathways akin to reasoning paths in large language models (LLMs). Specifically, inspired by chain-of-thought (CoT) reasoning in LLMs, we introduce the chain-of-reaction (CoR) notation that explicitly states reactants, reaction types, and intermediate products for each step in a pathway. With the CoR notation, ReaSyn can get dense supervision in every reaction step to explicitly learn chemical reaction rules during supervised training and perform step-by-step reasoning. In addition, to further enhance the reasoning capability of ReaSyn, we propose reinforcement learning (RL)-based finetuning and goal-directed test-time compute scaling tailored for synthesizable projection. ReaSyn achieves the highest reconstruction rate and pathway diversity in synthesizable molecule reconstruction and the highest optimization performance in synthesizable goal-directed molecular optimization, and significantly outperforms previous synthesizable projection methods in synthesizable hit expansion. These results highlight ReaSyn's superior ability to navigate combinatorially-large synthesizable chemical space.
GenMol: A Drug Discovery Generalist with Discrete Diffusion
Jan 10, 2025



Drug discovery is a complex process that involves multiple scenarios and stages, such as fragment-constrained molecule generation, hit generation and lead optimization. However, existing molecular generative models can only tackle one or two of these scenarios and lack the flexibility to address various aspects of the drug discovery pipeline. In this paper, we present Generalist Molecular generative model (GenMol), a versatile framework that addresses these limitations by applying discrete diffusion to the Sequential Attachment-based Fragment Embedding (SAFE) molecular representation. GenMol generates SAFE sequences through non-autoregressive bidirectional parallel decoding, thereby allowing utilization of a molecular context that does not rely on the specific token ordering and enhanced computational efficiency. Moreover, under the discrete diffusion framework, we introduce fragment remasking, a strategy that optimizes molecules by replacing fragments with masked tokens and regenerating them, enabling effective exploration of chemical space. GenMol significantly outperforms the previous GPT-based model trained on SAFE representations in de novo generation and fragment-constrained generation, and achieves state-of-the-art performance in goal-directed hit generation and lead optimization. These experimental results demonstrate that GenMol can tackle a wide range of drug discovery tasks, providing a unified and versatile approach for molecular design.
Molecule Generation with Fragment Retrieval Augmentation
Nov 18, 2024



Fragment-based drug discovery, in which molecular fragments are assembled into new molecules with desirable biochemical properties, has achieved great success. However, many fragment-based molecule generation methods show limited exploration beyond the existing fragments in the database as they only reassemble or slightly modify the given ones. To tackle this problem, we propose a new fragment-based molecule generation framework with retrieval augmentation, namely Fragment Retrieval-Augmented Generation (f-RAG). f-RAG is based on a pre-trained molecular generative model that proposes additional fragments from input fragments to complete and generate a new molecule. Given a fragment vocabulary, f-RAG retrieves two types of fragments: (1) hard fragments, which serve as building blocks that will be explicitly included in the newly generated molecule, and (2) soft fragments, which serve as reference to guide the generation of new fragments through a trainable fragment injection module. To extrapolate beyond the existing fragments, f-RAG updates the fragment vocabulary with generated fragments via an iterative refinement process which is further enhanced with post-hoc genetic fragment modification. f-RAG can achieve an improved exploration-exploitation trade-off by maintaining a pool of fragments and expanding it with novel and high-quality fragments through a strong generative prior.
BioNeMo Framework: a modular, high-performance library for AI model development in drug discovery
Nov 15, 2024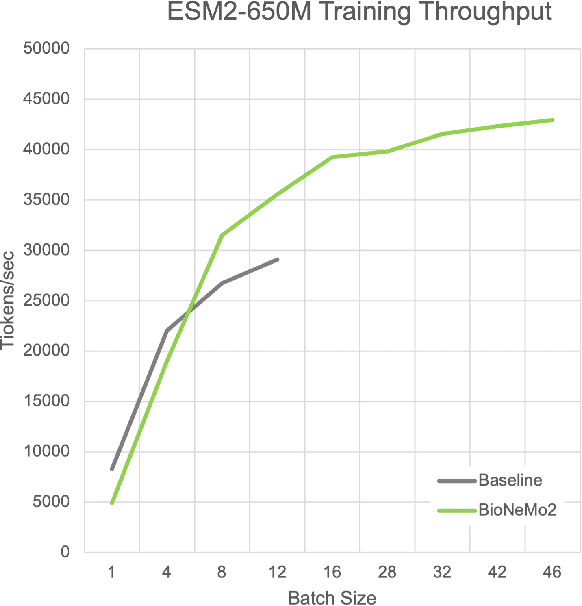
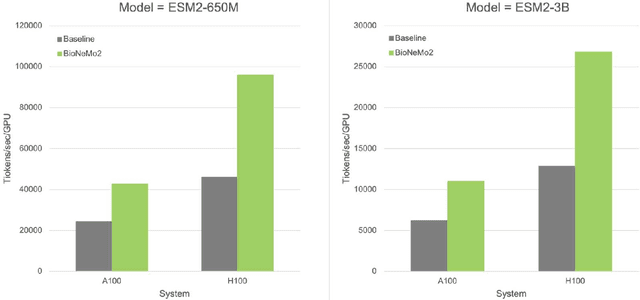
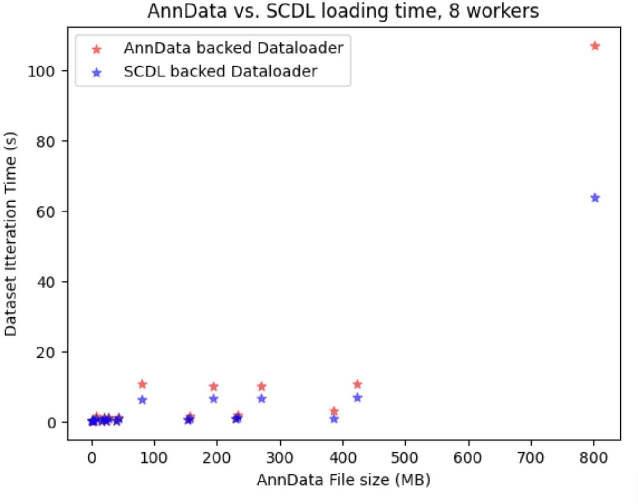
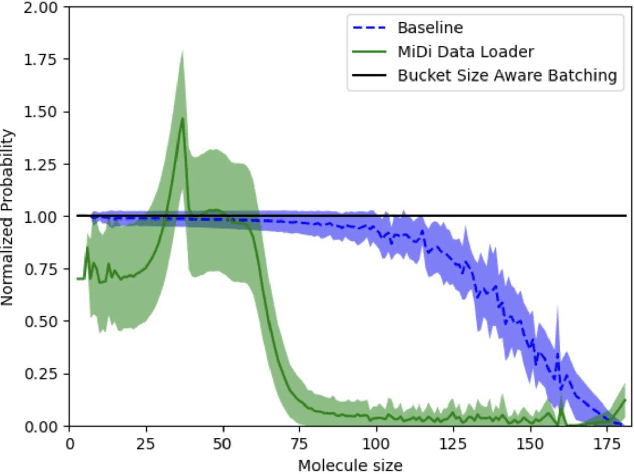
Artificial Intelligence models encoding biology and chemistry are opening new routes to high-throughput and high-quality in-silico drug development. However, their training increasingly relies on computational scale, with recent protein language models (pLM) training on hundreds of graphical processing units (GPUs). We introduce the BioNeMo Framework to facilitate the training of computational biology and chemistry AI models across hundreds of GPUs. Its modular design allows the integration of individual components, such as data loaders, into existing workflows and is open to community contributions. We detail technical features of the BioNeMo Framework through use cases such as pLM pre-training and fine-tuning. On 256 NVIDIA A100s, BioNeMo Framework trains a three billion parameter BERT-based pLM on over one trillion tokens in 4.2 days. The BioNeMo Framework is open-source and free for everyone to use.