 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRethinking Molecule Synthesizability with Chain-of-Reaction
Sep 19, 2025A well-known pitfall of molecular generative models is that they are not guaranteed to generate synthesizable molecules. There have been considerable attempts to address this problem, but given the exponentially large combinatorial space of synthesizable molecules, existing methods have shown limited coverage of the space and poor molecular optimization performance. To tackle these problems, we introduce ReaSyn, a generative framework for synthesizable projection where the model explores the neighborhood of given molecules in the synthesizable space by generating pathways that result in synthesizable analogs. To fully utilize the chemical knowledge contained in the synthetic pathways, we propose a novel perspective that views synthetic pathways akin to reasoning paths in large language models (LLMs). Specifically, inspired by chain-of-thought (CoT) reasoning in LLMs, we introduce the chain-of-reaction (CoR) notation that explicitly states reactants, reaction types, and intermediate products for each step in a pathway. With the CoR notation, ReaSyn can get dense supervision in every reaction step to explicitly learn chemical reaction rules during supervised training and perform step-by-step reasoning. In addition, to further enhance the reasoning capability of ReaSyn, we propose reinforcement learning (RL)-based finetuning and goal-directed test-time compute scaling tailored for synthesizable projection. ReaSyn achieves the highest reconstruction rate and pathway diversity in synthesizable molecule reconstruction and the highest optimization performance in synthesizable goal-directed molecular optimization, and significantly outperforms previous synthesizable projection methods in synthesizable hit expansion. These results highlight ReaSyn's superior ability to navigate combinatorially-large synthesizable chemical space.
GenMol: A Drug Discovery Generalist with Discrete Diffusion
Jan 10, 2025



Drug discovery is a complex process that involves multiple scenarios and stages, such as fragment-constrained molecule generation, hit generation and lead optimization. However, existing molecular generative models can only tackle one or two of these scenarios and lack the flexibility to address various aspects of the drug discovery pipeline. In this paper, we present Generalist Molecular generative model (GenMol), a versatile framework that addresses these limitations by applying discrete diffusion to the Sequential Attachment-based Fragment Embedding (SAFE) molecular representation. GenMol generates SAFE sequences through non-autoregressive bidirectional parallel decoding, thereby allowing utilization of a molecular context that does not rely on the specific token ordering and enhanced computational efficiency. Moreover, under the discrete diffusion framework, we introduce fragment remasking, a strategy that optimizes molecules by replacing fragments with masked tokens and regenerating them, enabling effective exploration of chemical space. GenMol significantly outperforms the previous GPT-based model trained on SAFE representations in de novo generation and fragment-constrained generation, and achieves state-of-the-art performance in goal-directed hit generation and lead optimization. These experimental results demonstrate that GenMol can tackle a wide range of drug discovery tasks, providing a unified and versatile approach for molecular design.
Molecule Generation with Fragment Retrieval Augmentation
Nov 18, 2024



Fragment-based drug discovery, in which molecular fragments are assembled into new molecules with desirable biochemical properties, has achieved great success. However, many fragment-based molecule generation methods show limited exploration beyond the existing fragments in the database as they only reassemble or slightly modify the given ones. To tackle this problem, we propose a new fragment-based molecule generation framework with retrieval augmentation, namely Fragment Retrieval-Augmented Generation (f-RAG). f-RAG is based on a pre-trained molecular generative model that proposes additional fragments from input fragments to complete and generate a new molecule. Given a fragment vocabulary, f-RAG retrieves two types of fragments: (1) hard fragments, which serve as building blocks that will be explicitly included in the newly generated molecule, and (2) soft fragments, which serve as reference to guide the generation of new fragments through a trainable fragment injection module. To extrapolate beyond the existing fragments, f-RAG updates the fragment vocabulary with generated fragments via an iterative refinement process which is further enhanced with post-hoc genetic fragment modification. f-RAG can achieve an improved exploration-exploitation trade-off by maintaining a pool of fragments and expanding it with novel and high-quality fragments through a strong generative prior.
Protein Representation Learning by Capturing Protein Sequence-Structure-Function Relationship
Apr 29, 2024



The goal of protein representation learning is to extract knowledge from protein databases that can be applied to various protein-related downstream tasks. Although protein sequence, structure, and function are the three key modalities for a comprehensive understanding of proteins, existing methods for protein representation learning have utilized only one or two of these modalities due to the difficulty of capturing the asymmetric interrelationships between them. To account for this asymmetry, we introduce our novel asymmetric multi-modal masked autoencoder (AMMA). AMMA adopts (1) a unified multi-modal encoder to integrate all three modalities into a unified representation space and (2) asymmetric decoders to ensure that sequence latent features reflect structural and functional information. The experiments demonstrate that the proposed AMMA is highly effective in learning protein representations that exhibit well-aligned inter-modal relationships, which in turn makes it effective for various downstream protein-related tasks.
A Simple and Scalable Representation for Graph Generation
Dec 04, 2023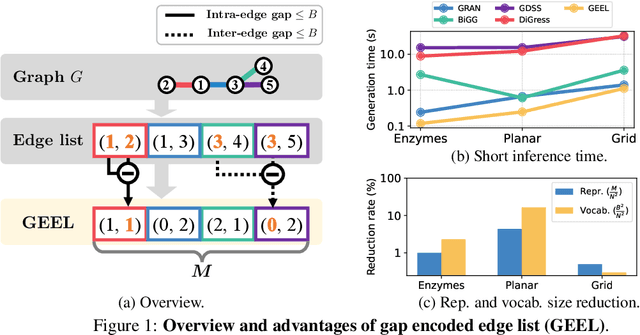
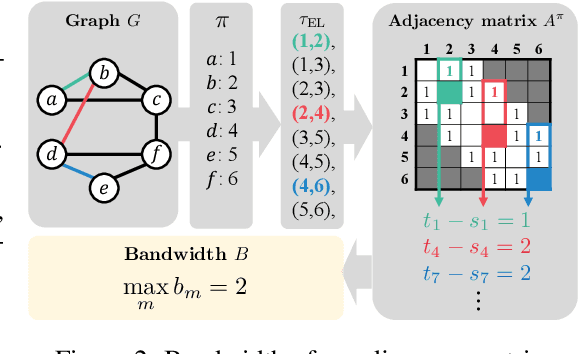
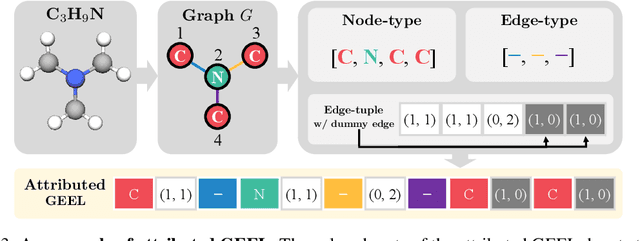
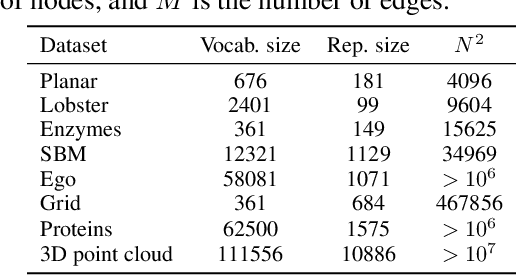
Recently, there has been a surge of interest in employing neural networks for graph generation, a fundamental statistical learning problem with critical applications like molecule design and community analysis. However, most approaches encounter significant limitations when generating large-scale graphs. This is due to their requirement to output the full adjacency matrices whose size grows quadratically with the number of nodes. In response to this challenge, we introduce a new, simple, and scalable graph representation named gap encoded edge list (GEEL) that has a small representation size that aligns with the number of edges. In addition, GEEL significantly reduces the vocabulary size by incorporating the gap encoding and bandwidth restriction schemes. GEEL can be autoregressively generated with the incorporation of node positional encoding, and we further extend GEEL to deal with attributed graphs by designing a new grammar. Our findings reveal that the adoption of this compact representation not only enhances scalability but also bolsters performance by simplifying the graph generation process. We conduct a comprehensive evaluation across ten non-attributed and two molecular graph generation tasks, demonstrating the effectiveness of GEEL.
Drug Discovery with Dynamic Goal-aware Fragments
Oct 02, 2023Fragment-based drug discovery is an effective strategy for discovering drug candidates in the vast chemical space, and has been widely employed in molecular generative models. However, many existing fragment extraction methods in such models do not take the target chemical properties into account or rely on heuristic rules. Additionally, the existing fragment-based generative models cannot update the fragment vocabulary with goal-aware fragments newly discovered during the generation. To this end, we propose a molecular generative framework for drug discovery, named Goal-aware fragment Extraction, Assembly, and Modification (GEAM). GEAM consists of three modules, each responsible for goal-aware fragment extraction, fragment assembly, and fragment modification. The fragment extraction module identifies important fragments that contribute to the desired target properties with the information bottleneck principle, thereby constructing an effective goal-aware fragment vocabulary. Moreover, GEAM can explore beyond the initial vocabulary with the fragment modification module, and the exploration is further enhanced through the dynamic goal-aware vocabulary update. We experimentally demonstrate that GEAM effectively discovers drug candidates through the generative cycle of the three modules in various drug discovery tasks.
Toward Automated Detection of Microbleeds with Anatomical Scale Localization: A Complete Clinical Diagnosis Support Using Deep Learning
Jun 22, 2023
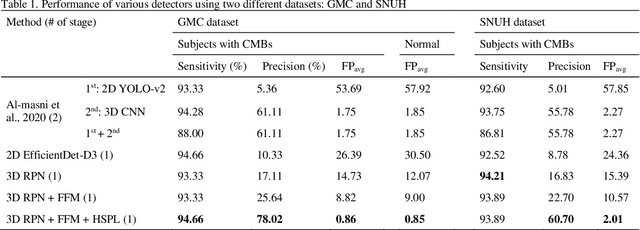
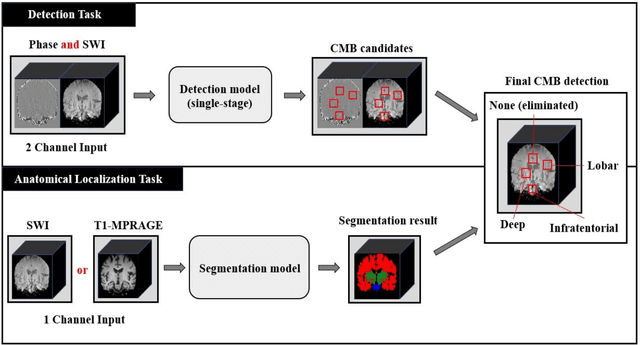
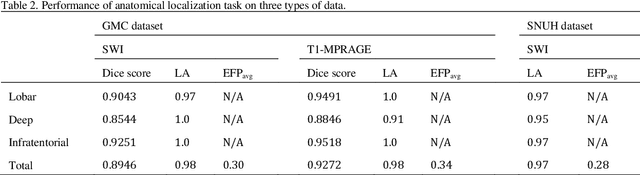
Cerebral Microbleeds (CMBs) are chronic deposits of small blood products in the brain tissues, which have explicit relation to various cerebrovascular diseases depending on their anatomical location, including cognitive decline, intracerebral hemorrhage, and cerebral infarction. However, manual detection of CMBs is a time-consuming and error-prone process because of their sparse and tiny structural properties. The detection of CMBs is commonly affected by the presence of many CMB mimics that cause a high false-positive rate (FPR), such as calcification and pial vessels. This paper proposes a novel 3D deep learning framework that does not only detect CMBs but also inform their anatomical location in the brain (i.e., lobar, deep, and infratentorial regions). For the CMB detection task, we propose a single end-to-end model by leveraging the U-Net as a backbone with Region Proposal Network (RPN). To significantly reduce the FPs within the same single model, we develop a new scheme, containing Feature Fusion Module (FFM) that detects small candidates utilizing contextual information and Hard Sample Prototype Learning (HSPL) that mines CMB mimics and generates additional loss term called concentration loss using Convolutional Prototype Learning (CPL). The anatomical localization task does not only tell to which region the CMBs belong but also eliminate some FPs from the detection task by utilizing anatomical information. The results show that the proposed RPN that utilizes the FFM and HSPL outperforms the vanilla RPN and achieves a sensitivity of 94.66% vs. 93.33% and an average number of false positives per subject (FPavg) of 0.86 vs. 14.73. Also, the anatomical localization task further improves the detection performance by reducing the FPavg to 0.56 while maintaining the sensitivity of 94.66%.
Score-based Generative Modeling of Graphs via the System of Stochastic Differential Equations
Feb 16, 2022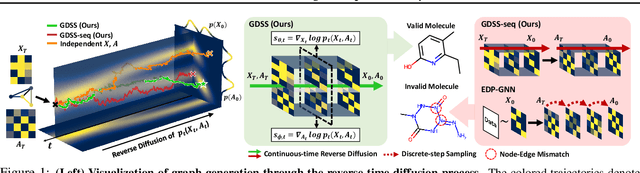

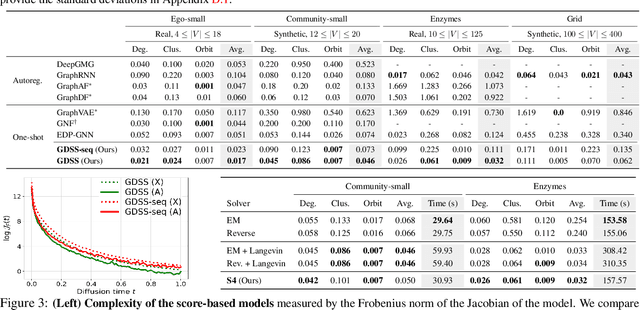
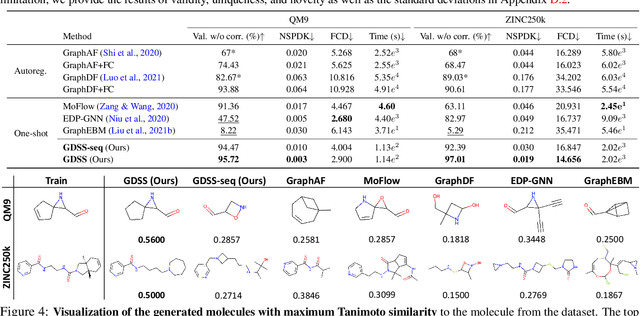
Generating graph-structured data requires learning the underlying distribution of graphs. Yet, this is a challenging problem, and the previous graph generative methods either fail to capture the permutation-invariance property of graphs or cannot sufficiently model the complex dependency between nodes and edges, which is crucial for generating real-world graphs such as molecules. To overcome such limitations, we propose a novel score-based generative model for graphs with a continuous-time framework. Specifically, we propose a new graph diffusion process that models the joint distribution of the nodes and edges through a system of stochastic differential equations (SDEs). Then, we derive novel score matching objectives tailored for the proposed diffusion process to estimate the gradient of the joint log-density with respect to each component, and introduce a new solver for the system of SDEs to efficiently sample from the reverse diffusion process. We validate our graph generation method on diverse datasets, on which it either achieves significantly superior or competitive performance to the baselines. Further analysis shows that our method is able to generate molecules that lie close to the training distribution yet do not violate the chemical valency rule, demonstrating the effectiveness of the system of SDEs in modeling the node-edge relationships.
Stacked U-Nets with Self-Assisted Priors Towards Robust Correction of Rigid Motion Artifact in Brain MRI
Nov 11, 2021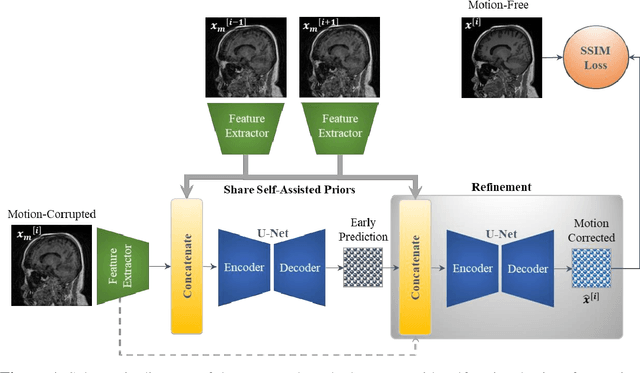

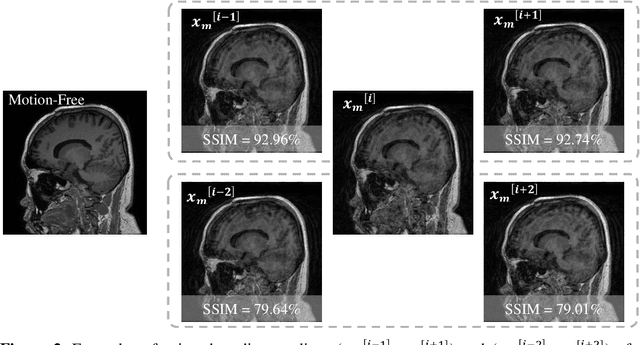
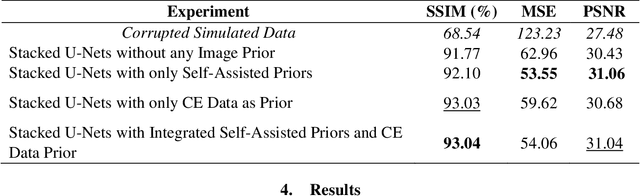
In this paper, we develop an efficient retrospective deep learning method called stacked U-Nets with self-assisted priors to address the problem of rigid motion artifacts in MRI. The proposed work exploits the usage of additional knowledge priors from the corrupted images themselves without the need for additional contrast data. The proposed network learns missed structural details through sharing auxiliary information from the contiguous slices of the same distorted subject. We further design a refinement stacked U-Nets that facilitates preserving of the image spatial details and hence improves the pixel-to-pixel dependency. To perform network training, simulation of MRI motion artifacts is inevitable. We present an intensive analysis using various types of image priors: the proposed self-assisted priors and priors from other image contrast of the same subject. The experimental analysis proves the effectiveness and feasibility of our self-assisted priors since it does not require any further data scans.
Hit and Lead Discovery with Explorative RL and Fragment-based Molecule Generation
Oct 05, 2021
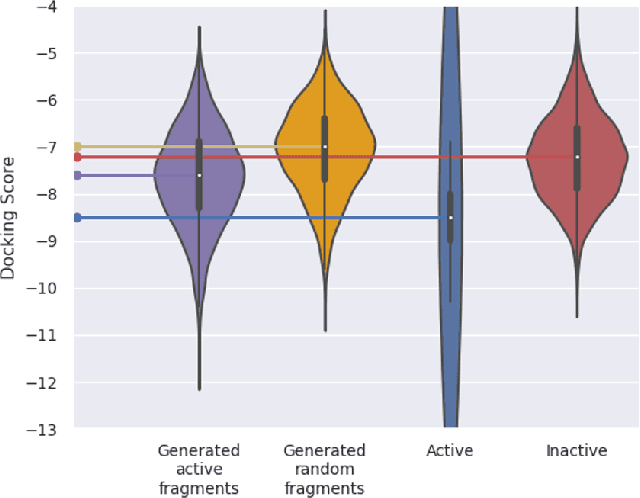

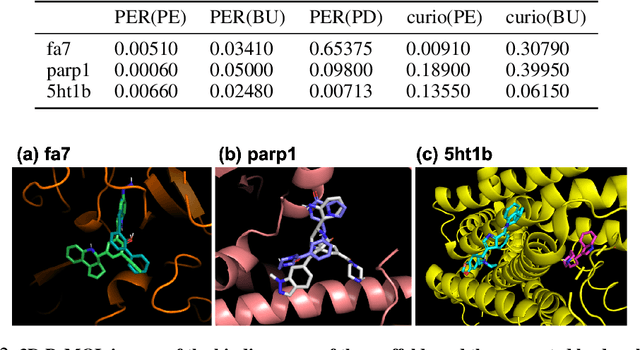
Recently, utilizing reinforcement learning (RL) to generate molecules with desired properties has been highlighted as a promising strategy for drug design. A molecular docking program - a physical simulation that estimates protein-small molecule binding affinity - can be an ideal reward scoring function for RL, as it is a straightforward proxy of the therapeutic potential. Still, two imminent challenges exist for this task. First, the models often fail to generate chemically realistic and pharmacochemically acceptable molecules. Second, the docking score optimization is a difficult exploration problem that involves many local optima and less smooth surfaces with respect to molecular structure. To tackle these challenges, we propose a novel RL framework that generates pharmacochemically acceptable molecules with large docking scores. Our method - Fragment-based generative RL with Explorative Experience replay for Drug design (FREED) - constrains the generated molecules to a realistic and qualified chemical space and effectively explores the space to find drugs by coupling our fragment-based generation method and a novel error-prioritized experience replay (PER). We also show that our model performs well on both de novo and scaffold-based schemes. Our model produces molecules of higher quality compared to existing methods while achieving state-of-the-art performance on two of three targets in terms of the docking scores of the generated molecules. We further show with ablation studies that our method, predictive error-PER (FREED(PE)), significantly improves the model performance.