 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScore-informed Neural Operator for Enhancing Ordering-based Causal Discovery
Aug 18, 2025Ordering-based approaches to causal discovery identify topological orders of causal graphs, providing scalable alternatives to combinatorial search methods. Under the Additive Noise Model (ANM) assumption, recent causal ordering methods based on score matching require an accurate estimation of the Hessian diagonal of the log-densities. However, previous approaches mainly use Stein gradient estimators, which are computationally expensive and memory-intensive. Although DiffAN addresses these limitations by substituting kernel-based estimates with diffusion models, it remains numerically unstable due to the second-order derivatives of score models. To alleviate these problems, we propose Score-informed Neural Operator (SciNO), a probabilistic generative model in smooth function spaces designed to stably approximate the Hessian diagonal and to preserve structural information during the score modeling. Empirical results show that SciNO reduces order divergence by 42.7% on synthetic graphs and by 31.5% on real-world datasets on average compared to DiffAN, while maintaining memory efficiency and scalability. Furthermore, we propose a probabilistic control algorithm for causal reasoning with autoregressive models that integrates SciNO's probability estimates with autoregressive model priors, enabling reliable data-driven causal ordering informed by semantic information. Consequently, the proposed method enhances causal reasoning abilities of LLMs without additional fine-tuning or prompt engineering.
Hit and Lead Discovery with Explorative RL and Fragment-based Molecule Generation
Oct 05, 2021
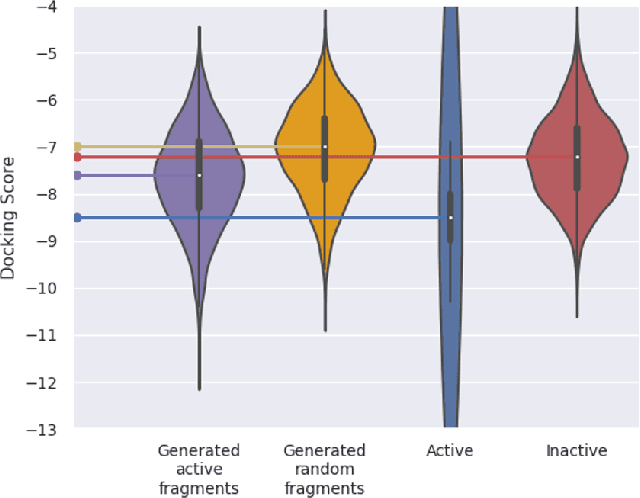

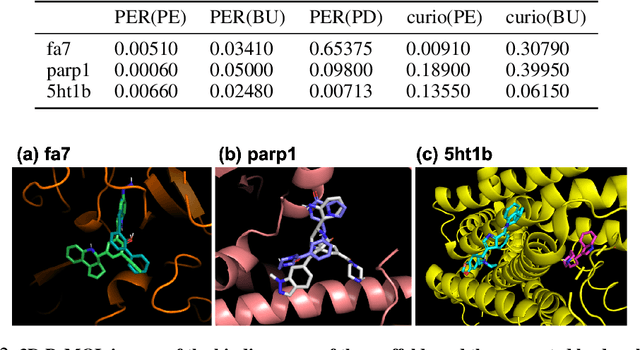
Recently, utilizing reinforcement learning (RL) to generate molecules with desired properties has been highlighted as a promising strategy for drug design. A molecular docking program - a physical simulation that estimates protein-small molecule binding affinity - can be an ideal reward scoring function for RL, as it is a straightforward proxy of the therapeutic potential. Still, two imminent challenges exist for this task. First, the models often fail to generate chemically realistic and pharmacochemically acceptable molecules. Second, the docking score optimization is a difficult exploration problem that involves many local optima and less smooth surfaces with respect to molecular structure. To tackle these challenges, we propose a novel RL framework that generates pharmacochemically acceptable molecules with large docking scores. Our method - Fragment-based generative RL with Explorative Experience replay for Drug design (FREED) - constrains the generated molecules to a realistic and qualified chemical space and effectively explores the space to find drugs by coupling our fragment-based generation method and a novel error-prioritized experience replay (PER). We also show that our model performs well on both de novo and scaffold-based schemes. Our model produces molecules of higher quality compared to existing methods while achieving state-of-the-art performance on two of three targets in terms of the docking scores of the generated molecules. We further show with ablation studies that our method, predictive error-PER (FREED(PE)), significantly improves the model performance.
A benchmark study on reliable molecular supervised learning via Bayesian learning
Jul 01, 2020

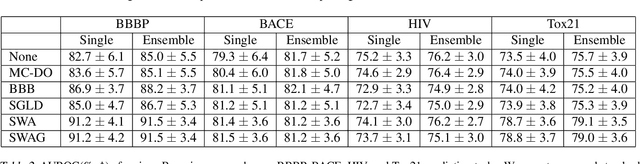

Virtual screening aims to find desirable compounds from chemical library by using computational methods. For this purpose with machine learning, model outputs that can be interpreted as predictive probability will be beneficial, in that a high prediction score corresponds to high probability of correctness. In this work, we present a study on the prediction performance and reliability of graph neural networks trained with the recently proposed Bayesian learning algorithms. Our work shows that Bayesian learning algorithms allow well-calibrated predictions for various GNN architectures and classification tasks. Also, we show the implications of reliable predictions on virtual screening, where Bayesian learning may lead to higher success in finding hit compounds.