 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHit and Lead Discovery with Explorative RL and Fragment-based Molecule Generation
Paper and Code

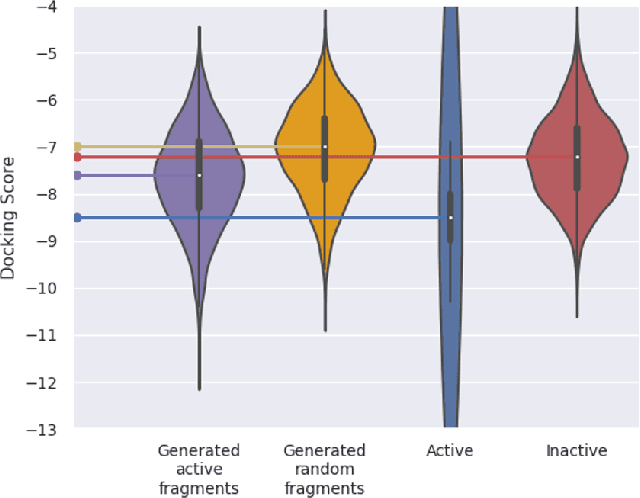

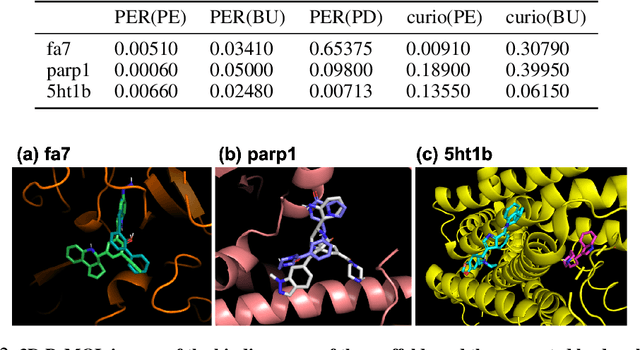
Recently, utilizing reinforcement learning (RL) to generate molecules with desired properties has been highlighted as a promising strategy for drug design. A molecular docking program - a physical simulation that estimates protein-small molecule binding affinity - can be an ideal reward scoring function for RL, as it is a straightforward proxy of the therapeutic potential. Still, two imminent challenges exist for this task. First, the models often fail to generate chemically realistic and pharmacochemically acceptable molecules. Second, the docking score optimization is a difficult exploration problem that involves many local optima and less smooth surfaces with respect to molecular structure. To tackle these challenges, we propose a novel RL framework that generates pharmacochemically acceptable molecules with large docking scores. Our method - Fragment-based generative RL with Explorative Experience replay for Drug design (FREED) - constrains the generated molecules to a realistic and qualified chemical space and effectively explores the space to find drugs by coupling our fragment-based generation method and a novel error-prioritized experience replay (PER). We also show that our model performs well on both de novo and scaffold-based schemes. Our model produces molecules of higher quality compared to existing methods while achieving state-of-the-art performance on two of three targets in terms of the docking scores of the generated molecules. We further show with ablation studies that our method, predictive error-PER (FREED(PE)), significantly improves the model performance.