 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Priori Sampling of Transition States with Guided Diffusion
Mar 26, 2026Transition states, the first-order saddle points on the potential energy surfaces, govern the kinetics and mechanisms of chemical reactions and conformational changes. Locating them is challenging because transition pathways are topologically complex and can proceed via an ensemble of diverse routes. Existing methods address these challenges by introducing heuristic assumptions about the pathway or reaction coordinates, which limits their applicability when a good initial guess is unavailable or when the guess precludes alternative, potentially relevant pathways. We propose to bypass such heuristic limitations by introducing ASTRA, A Priori Sampling of TRAnsition States with Guided Diffusion, which reframes the transition state search as an inference-time scaling problem for generative models. ASTRA trains a score-based diffusion model on configurations from known metastable states. Then, ASTRA guides inference toward the isodensity surface separating the basins of metastable states via a principled composition of conditional scores. A Score-Aligned Ascent (SAA) process then approximates a reaction coordinate from the difference between conditioned scores and combines it with physical forces to drive convergence onto first-order transition states. Validated on benchmarks from 2D potentials to biomolecular conformational changes and chemical reaction, ASTRA locates transition states with high precision and discovers multiple reaction pathways, enabling mechanistic studies of complex molecular systems.
Learning Collective Variables from Time-lagged Generation
Jul 10, 2025Rare events such as state transitions are difficult to observe directly with molecular dynamics simulations due to long timescales. Enhanced sampling techniques overcome this by introducing biases along carefully chosen low-dimensional features, known as collective variables (CVs), which capture the slow degrees of freedom. Machine learning approaches (MLCVs) have automated CV discovery, but existing methods typically focus on discriminating meta-stable states without fully encoding the detailed dynamics essential for accurate sampling. We propose TLC, a framework that learns CVs directly from time-lagged conditions of a generative model. Instead of modeling the static Boltzmann distribution, TLC models a time-lagged conditional distribution yielding CVs to capture the slow dynamic behavior. We validate TLC on the Alanine Dipeptide system using two CV-based enhanced sampling tasks: (i) steered molecular dynamics (SMD) and (ii) on-the-fly probability enhanced sampling (OPES), demonstrating equal or superior performance compared to existing MLCV methods in both transition path sampling and state discrimination.
Transferable Learning of Reaction Pathways from Geometric Priors
Apr 21, 2025



Identifying minimum-energy paths (MEPs) is crucial for understanding chemical reaction mechanisms but remains computationally demanding. We introduce MEPIN, a scalable machine-learning method for efficiently predicting MEPs from reactant and product configurations, without relying on transition-state geometries or pre-optimized reaction paths during training. The task is defined as predicting deviations from geometric interpolations along reaction coordinates. We address this task with a continuous reaction path model based on a symmetry-broken equivariant neural network that generates a flexible number of intermediate structures. The model is trained using an energy-based objective, with efficiency enhanced by incorporating geometric priors from geodesic interpolation as initial interpolations or pre-training objectives. Our approach generalizes across diverse chemical reactions and achieves accurate alignment with reference intrinsic reaction coordinates, as demonstrated on various small molecule reactions and [3+2] cycloadditions. Our method enables the exploration of large chemical reaction spaces with efficient, data-driven predictions of reaction pathways.
Flow Matching for Accelerated Simulation of Atomic Transport in Materials
Oct 02, 2024We introduce LiFlow, a generative framework to accelerate molecular dynamics (MD) simulations for crystalline materials that formulates the task as conditional generation of atomic displacements. The model uses flow matching, with a Propagator submodel to generate atomic displacements and a Corrector to locally correct unphysical geometries, and incorporates an adaptive prior based on the Maxwell-Boltzmann distribution to account for chemical and thermal conditions. We benchmark LiFlow on a dataset comprising 25-ps trajectories of lithium diffusion across 4,186 solid-state electrolyte (SSE) candidates at four temperatures. The model obtains a consistent Spearman rank correlation of 0.7-0.8 for lithium mean squared displacement (MSD) predictions on unseen compositions. Furthermore, LiFlow generalizes from short training trajectories to larger supercells and longer simulations while maintaining high accuracy. With speed-ups of up to 600,000$\times$ compared to first-principles methods, LiFlow enables scalable simulations at significantly larger length and time scales.
Learning Collective Variables for Protein Folding with Labeled Data Augmentation through Geodesic Interpolation
Feb 02, 2024



In molecular dynamics (MD) simulations, rare events, such as protein folding, are typically studied by means of enhanced sampling techniques, most of which rely on the definition of a collective variable (CV) along which the acceleration occurs. Obtaining an expressive CV is crucial, but often hindered by the lack of information about the particular event, e.g., the transition from unfolded to folded conformation. We propose a simulation-free data augmentation strategy using physics-inspired metrics to generate geodesic interpolations resembling protein folding transitions, thereby improving sampling efficiency without true transition state samples. Leveraging interpolation progress parameters, we introduce a regression-based learning scheme for CV models, which outperforms classifier-based methods when transition state data is limited and noisy
Chemically Transferable Generative Backmapping of Coarse-Grained Proteins
Mar 02, 2023Coarse-graining (CG) accelerates molecular simulations of protein dynamics by simulating sets of atoms as singular beads. Backmapping is the opposite operation of bringing lost atomistic details back from the CG representation. While machine learning (ML) has produced accurate and efficient CG simulations of proteins, fast and reliable backmapping remains a challenge. Rule-based methods produce poor all-atom geometries, needing computationally costly refinement through additional simulations. Recently proposed ML approaches outperform traditional baselines but are not transferable between proteins and sometimes generate unphysical atom placements with steric clashes and implausible torsion angles. This work addresses both issues to build a fast, transferable, and reliable generative backmapping tool for CG protein representations. We achieve generalization and reliability through a combined set of innovations: representation based on internal coordinates; an equivariant encoder/prior; a custom loss function that helps ensure local structure, global structure, and physical constraints; and expert curation of high-quality out-of-equilibrium protein data for training. Our results pave the way for out-of-the-box backmapping of coarse-grained simulations for arbitrary proteins.
Hit and Lead Discovery with Explorative RL and Fragment-based Molecule Generation
Oct 05, 2021
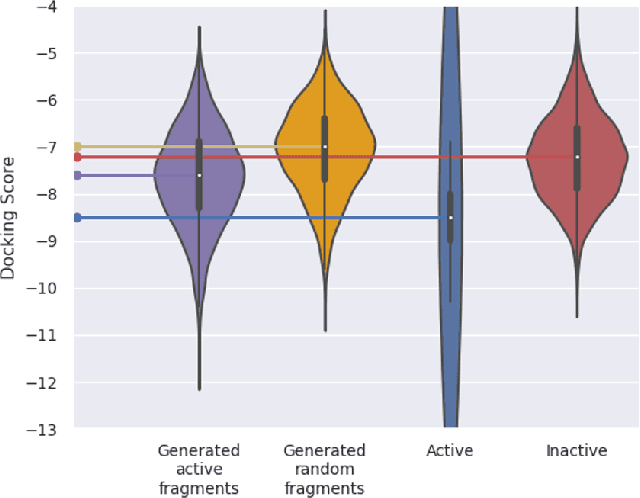

Recently, utilizing reinforcement learning (RL) to generate molecules with desired properties has been highlighted as a promising strategy for drug design. A molecular docking program - a physical simulation that estimates protein-small molecule binding affinity - can be an ideal reward scoring function for RL, as it is a straightforward proxy of the therapeutic potential. Still, two imminent challenges exist for this task. First, the models often fail to generate chemically realistic and pharmacochemically acceptable molecules. Second, the docking score optimization is a difficult exploration problem that involves many local optima and less smooth surfaces with respect to molecular structure. To tackle these challenges, we propose a novel RL framework that generates pharmacochemically acceptable molecules with large docking scores. Our method - Fragment-based generative RL with Explorative Experience replay for Drug design (FREED) - constrains the generated molecules to a realistic and qualified chemical space and effectively explores the space to find drugs by coupling our fragment-based generation method and a novel error-prioritized experience replay (PER). We also show that our model performs well on both de novo and scaffold-based schemes. Our model produces molecules of higher quality compared to existing methods while achieving state-of-the-art performance on two of three targets in terms of the docking scores of the generated molecules. We further show with ablation studies that our method, predictive error-PER (FREED(PE)), significantly improves the model performance.
PIGNet: A physics-informed deep learning model toward generalized drug-target interaction predictions
Aug 22, 2020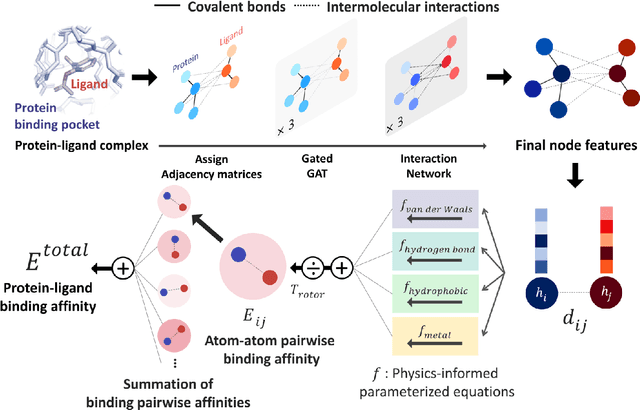
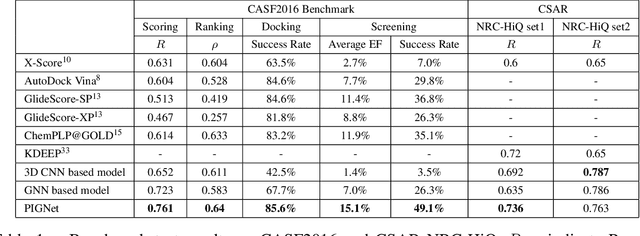
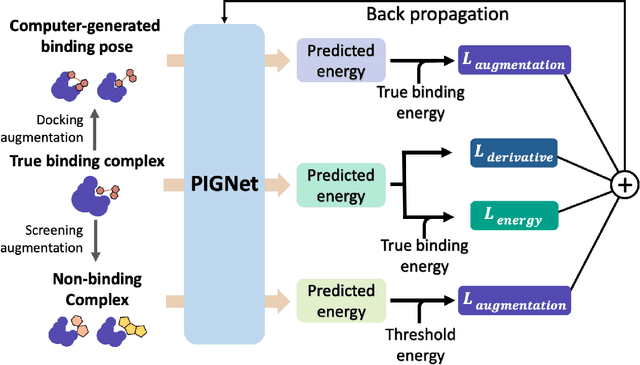
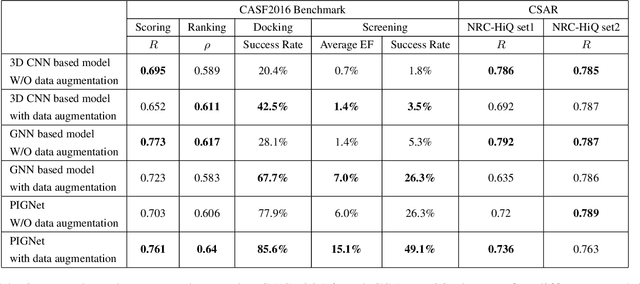
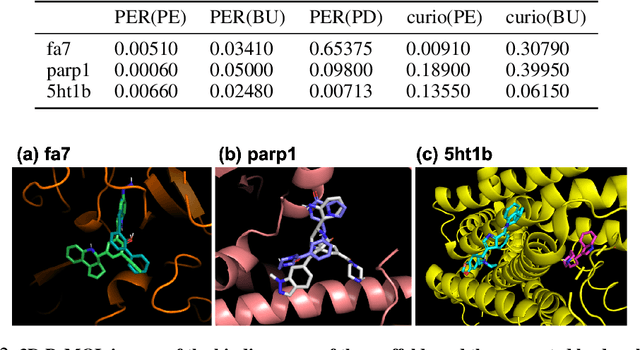
Recently, deep neural network (DNN)-based drug-target interaction (DTI) models are highlighted for their high accuracy with affordable computational costs. Yet, the models' insufficient generalization remains a challenging problem in the practice of in-silico drug discovery. We propose two key strategies to enhance generalization in the DTI model. The first one is to integrate physical models into DNN models. Our model, PIGNet, predicts the atom-atom pairwise interactions via physics-informed equations parameterized with neural networks and provides the total binding affinity of a protein-ligand complex as their sum. We further improved the model generalization by augmenting a wider range of binding poses and ligands to training data. PIGNet achieved a significant improvement in docking success rate, screening enhancement factor, and screening success rate by up to 2.01, 10.78, 14.0 times, respectively, compared to the previous DNN models. The physics-informed model also enables the interpretation of predicted binding affinities by visualizing the energy contribution of ligand substructures, providing insights for ligand optimization. Finally, we devised the uncertainty estimator of our model's prediction to qualify the outcomes and reduce the false positive rates.
A comprehensive study on the prediction reliability of graph neural networks for virtual screening
Mar 17, 2020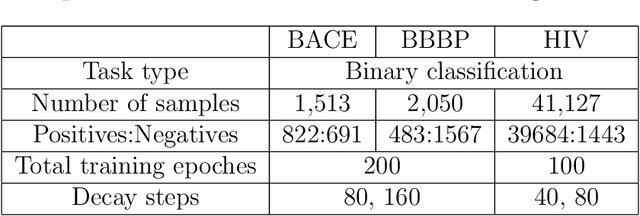
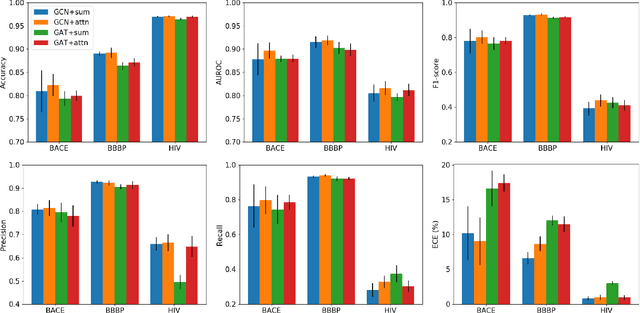
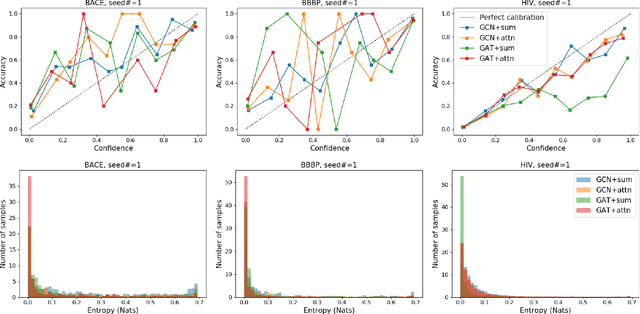
Prediction models based on deep neural networks are increasingly gaining attention for fast and accurate virtual screening systems. For decision makings in virtual screening, researchers find it useful to interpret an output of classification system as probability, since such interpretation allows them to filter out more desirable compounds. However, probabilistic interpretation cannot be correct for models that hold over-parameterization problems or inappropriate regularizations, leading to unreliable prediction and decision making. In this regard, we concern the reliability of neural prediction models on molecular properties, especially when models are trained with sparse data points and imbalanced distributions. This work aims to propose guidelines for training reliable models, we thus provide methodological details and ablation studies on the following train principles. We investigate the effects of model architectures, regularization methods, and loss functions on the prediction performance and reliability of classification results. Moreover, we evaluate prediction reliability of models on virtual screening scenario. Our result highlights that correct choice of regularization and inference methods is evidently important to achieve high success rate, especially in data imbalanced situation. All experiments were performed under a single unified model implementation to alleviate external randomness in model training and to enable precise comparison of results.