 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAtom-level Protein Representation Learning Improves Protein Structure Prediction
May 21, 2026Recent advances in generative modeling show that pretrained representations can improve generation as conditioning features or alignment targets. Motivated by this, we study protein representations for predicting structures beyond conventional function annotation. We propose TriProRep, a structure-aware pretraining method that jointly models three aligned residue-level views: amino-acid identity, backbone geometry, and local full-atom geometry, discretely encoded via VQ-VAE tokenizers. By pretraining to recover original tokens from generator-corrupted views, TriProRep learns to distinguish plausible but incorrect cross-view augmentations from the original protein. We further introduce RepSP, a benchmark for evaluating protein representations in structure-predictive settings. RepSP tests three uses of representations: homodimer co-folding from apo-chain representations, residue-level prediction of homodimer-derived interaction properties, and representation-aligned monomer structure prediction. Across these tasks, TriProRep improves over sequence-only and prior structure-aware representation models, while maintaining competitive performance on conventional benchmarks.
NCIDiff: Non-covalent Interaction-generative Diffusion Model for Improving Reliability of 3D Molecule Generation Inside Protein Pocket
May 27, 2024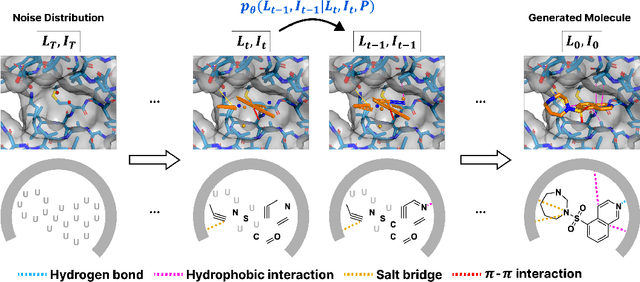
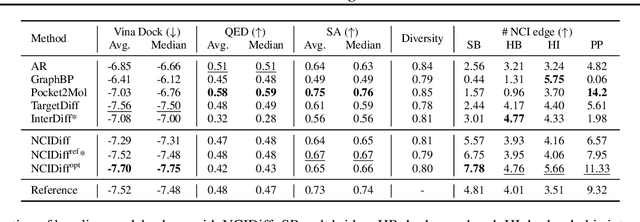
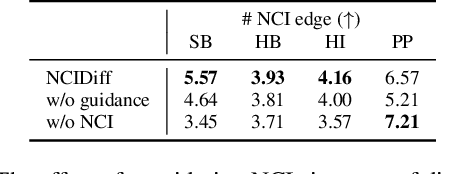
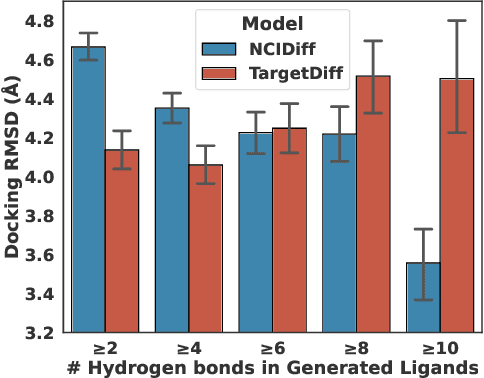
Advancements in deep generative modeling have changed the paradigm of drug discovery. Among such approaches, target-aware methods that exploit 3D structures of protein pockets were spotlighted for generating ligand molecules with their plausible binding modes. While docking scores superficially assess the quality of generated ligands, closer inspection of the binding structures reveals the inconsistency in local interactions between a pocket and generated ligands. Here, we address the issue by explicitly generating non-covalent interactions (NCIs), which are universal patterns throughout protein-ligand complexes. Our proposed model, NCIDiff, simultaneously denoises NCI types of protein-ligand edges along with a 3D graph of a ligand molecule during the sampling. With the NCI-generating strategy, our model generates ligands with more reliable NCIs, especially outperforming the baseline diffusion-based models. We further adopted inpainting techniques on NCIs to further improve the quality of the generated molecules. Finally, we showcase the applicability of NCIDiff on drug design tasks for real-world settings with specialized objectives by guiding the generation process with desired NCI patterns.
PIGNet: A physics-informed deep learning model toward generalized drug-target interaction predictions
Aug 22, 2020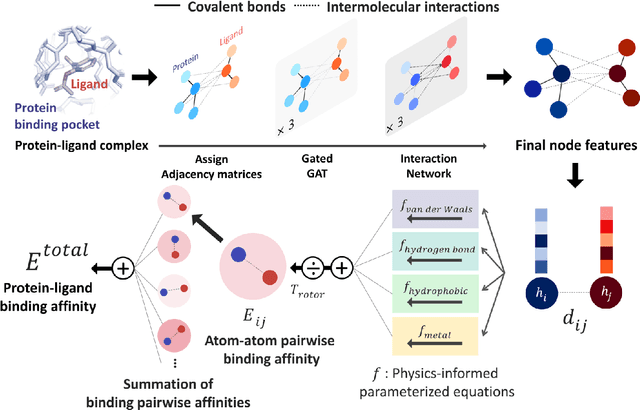
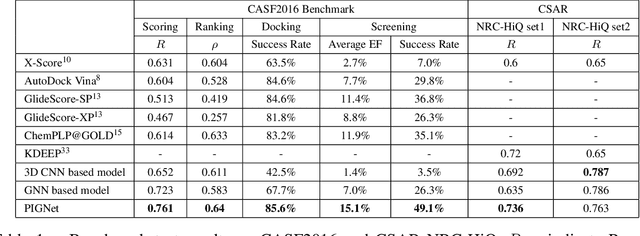
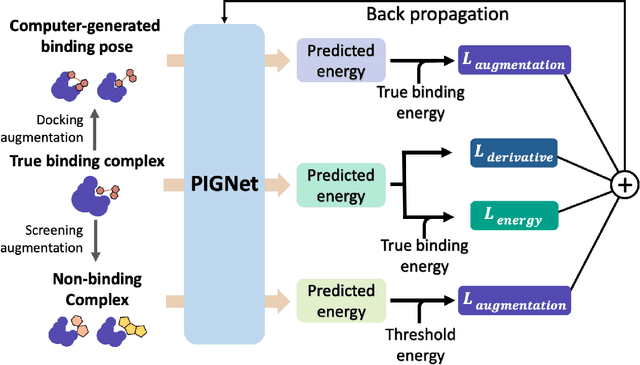
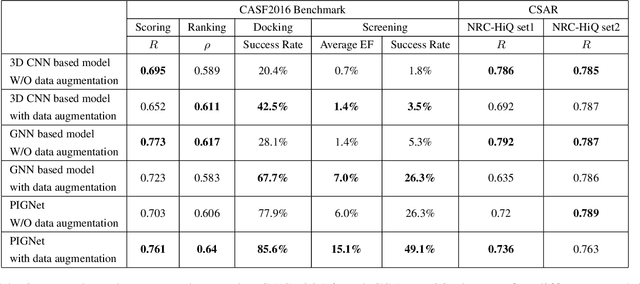
Recently, deep neural network (DNN)-based drug-target interaction (DTI) models are highlighted for their high accuracy with affordable computational costs. Yet, the models' insufficient generalization remains a challenging problem in the practice of in-silico drug discovery. We propose two key strategies to enhance generalization in the DTI model. The first one is to integrate physical models into DNN models. Our model, PIGNet, predicts the atom-atom pairwise interactions via physics-informed equations parameterized with neural networks and provides the total binding affinity of a protein-ligand complex as their sum. We further improved the model generalization by augmenting a wider range of binding poses and ligands to training data. PIGNet achieved a significant improvement in docking success rate, screening enhancement factor, and screening success rate by up to 2.01, 10.78, 14.0 times, respectively, compared to the previous DNN models. The physics-informed model also enables the interpretation of predicted binding affinities by visualizing the energy contribution of ligand substructures, providing insights for ligand optimization. Finally, we devised the uncertainty estimator of our model's prediction to qualify the outcomes and reduce the false positive rates.