 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeneral Binding Affinity Guidance for Diffusion Models in Structure-Based Drug Design
Paper and Code
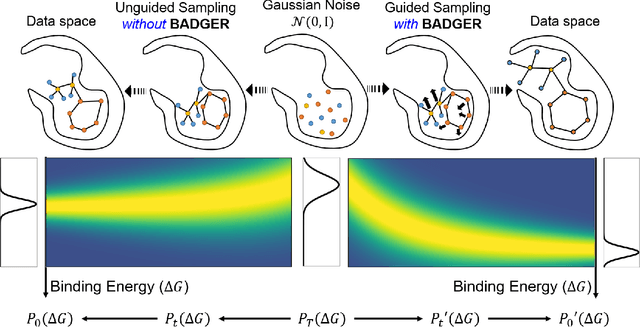
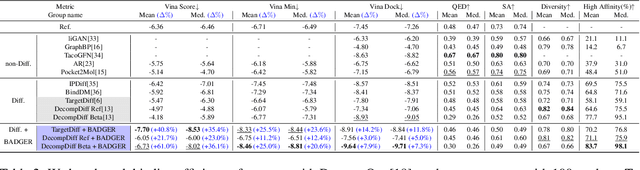
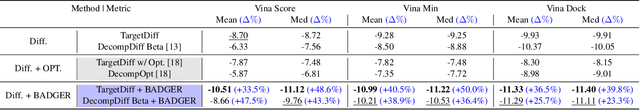
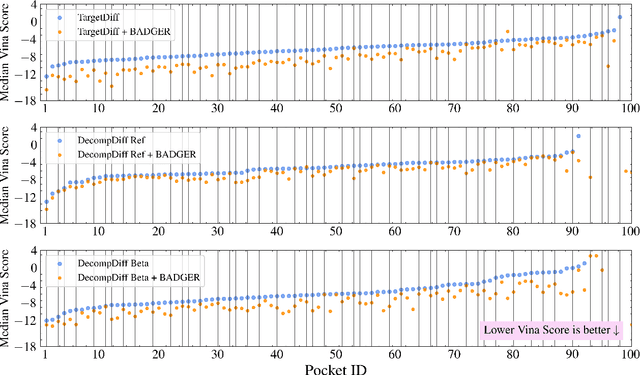
Structure-Based Drug Design (SBDD) focuses on generating valid ligands that strongly and specifically bind to a designated protein pocket. Several methods use machine learning for SBDD to generate these ligands in 3D space, conditioned on the structure of a desired protein pocket. Recently, diffusion models have shown success here by modeling the underlying distributions of atomic positions and types. While these methods are effective in considering the structural details of the protein pocket, they often fail to explicitly consider the binding affinity. Binding affinity characterizes how tightly the ligand binds to the protein pocket, and is measured by the change in free energy associated with the binding process. It is one of the most crucial metrics for benchmarking the effectiveness of the interaction between a ligand and protein pocket. To address this, we propose BADGER: Binding Affinity Diffusion Guidance with Enhanced Refinement. BADGER is a general guidance method to steer the diffusion sampling process towards improved protein-ligand binding, allowing us to adjust the distribution of the binding affinity between ligands and proteins. Our method is enabled by using a neural network (NN) to model the energy function, which is commonly approximated by AutoDock Vina (ADV). ADV's energy function is non-differentiable, and estimates the affinity based on the interactions between a ligand and target protein receptor. By using a NN as a differentiable energy function proxy, we utilize the gradient of our learned energy function as a guidance method on top of any trained diffusion model. We show that our method improves the binding affinity of generated ligands to their protein receptors by up to 60\%, significantly surpassing previous machine learning methods. We also show that our guidance method is flexible and can be easily applied to other diffusion-based SBDD frameworks.