 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeneral Binding Affinity Guidance for Diffusion Models in Structure-Based Drug Design
Jun 24, 2024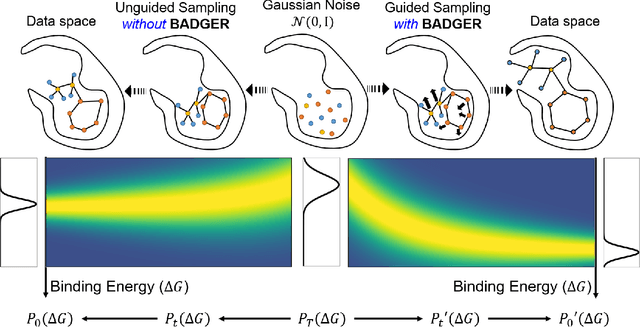
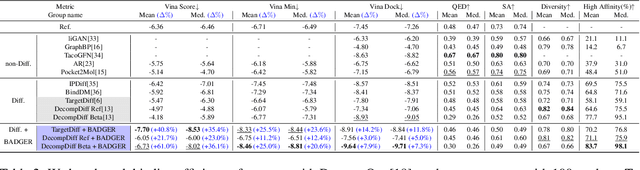
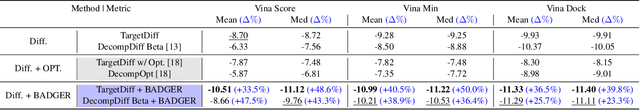
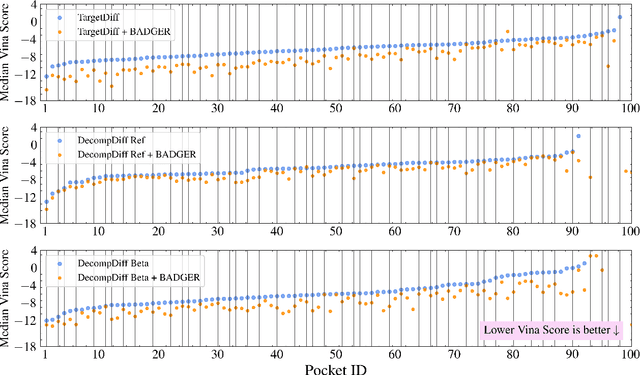
Structure-Based Drug Design (SBDD) focuses on generating valid ligands that strongly and specifically bind to a designated protein pocket. Several methods use machine learning for SBDD to generate these ligands in 3D space, conditioned on the structure of a desired protein pocket. Recently, diffusion models have shown success here by modeling the underlying distributions of atomic positions and types. While these methods are effective in considering the structural details of the protein pocket, they often fail to explicitly consider the binding affinity. Binding affinity characterizes how tightly the ligand binds to the protein pocket, and is measured by the change in free energy associated with the binding process. It is one of the most crucial metrics for benchmarking the effectiveness of the interaction between a ligand and protein pocket. To address this, we propose BADGER: Binding Affinity Diffusion Guidance with Enhanced Refinement. BADGER is a general guidance method to steer the diffusion sampling process towards improved protein-ligand binding, allowing us to adjust the distribution of the binding affinity between ligands and proteins. Our method is enabled by using a neural network (NN) to model the energy function, which is commonly approximated by AutoDock Vina (ADV). ADV's energy function is non-differentiable, and estimates the affinity based on the interactions between a ligand and target protein receptor. By using a NN as a differentiable energy function proxy, we utilize the gradient of our learned energy function as a guidance method on top of any trained diffusion model. We show that our method improves the binding affinity of generated ligands to their protein receptors by up to 60\%, significantly surpassing previous machine learning methods. We also show that our guidance method is flexible and can be easily applied to other diffusion-based SBDD frameworks.
Predicting CO$_2$ Absorption in Ionic Liquids with Molecular Descriptors and Explainable Graph Neural Networks
Sep 29, 2022
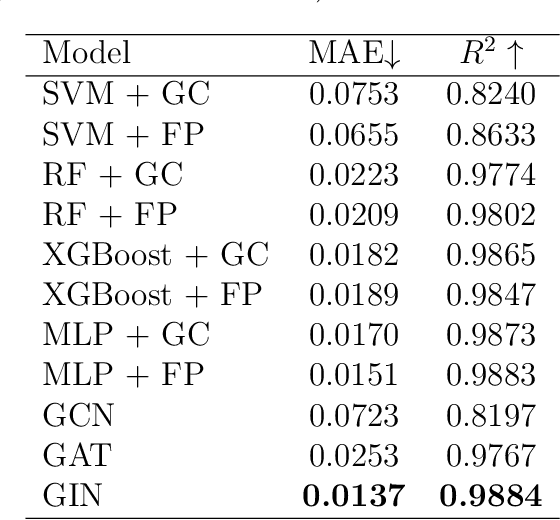

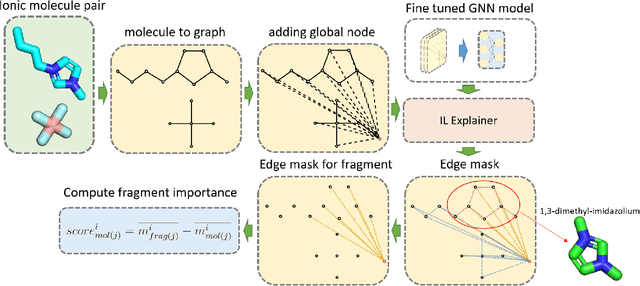
Ionic Liquids (ILs) provide a promising solution for CO$_2$ capture and storage to mitigate global warming. However, identifying and designing the high-capacity IL from the giant chemical space requires expensive, and exhaustive simulations and experiments. Machine learning (ML) can accelerate the process of searching for desirable ionic molecules through accurate and efficient property predictions in a data-driven manner. But existing descriptors and ML models for the ionic molecule suffer from the inefficient adaptation of molecular graph structure. Besides, few works have investigated the explainability of ML models to help understand the learned features that can guide the design of efficient ionic molecules. In this work, we develop both fingerprint-based ML models and Graph Neural Networks (GNNs) to predict the CO$_2$ absorption in ILs. Fingerprint works on graph structure at the feature extraction stage, while GNNs directly handle molecule structure in both the feature extraction and model prediction stage. We show that our method outperforms previous ML models by reaching a high accuracy (MAE of 0.0137, $R^2$ of 0.9884). Furthermore, we take the advantage of GNNs feature representation and develop a substructure-based explanation method that provides insight into how each chemical fragments within IL molecules contribute to the CO$_2$ absorption prediction of ML models. We also show that our explanation result agrees with some ground truth from the theoretical reaction mechanism of CO$_2$ absorption in ILs, which can advise on the design of novel and efficient functional ILs in the future.