 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting CO$_2$ Absorption in Ionic Liquids with Molecular Descriptors and Explainable Graph Neural Networks
Paper and Code

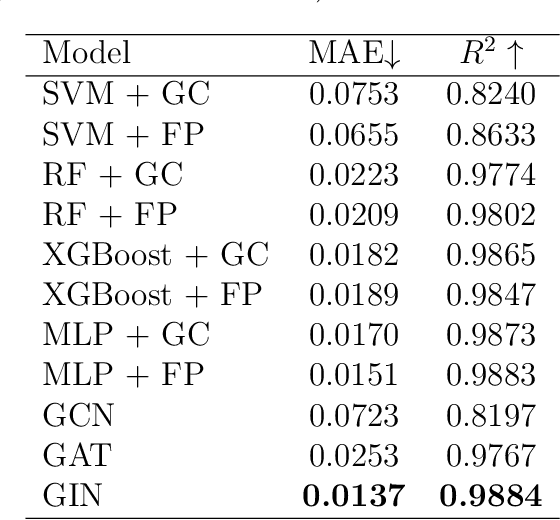

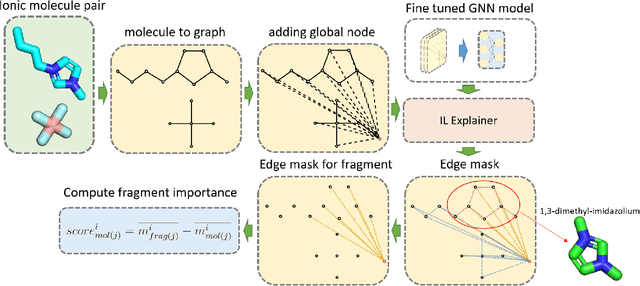
Ionic Liquids (ILs) provide a promising solution for CO$_2$ capture and storage to mitigate global warming. However, identifying and designing the high-capacity IL from the giant chemical space requires expensive, and exhaustive simulations and experiments. Machine learning (ML) can accelerate the process of searching for desirable ionic molecules through accurate and efficient property predictions in a data-driven manner. But existing descriptors and ML models for the ionic molecule suffer from the inefficient adaptation of molecular graph structure. Besides, few works have investigated the explainability of ML models to help understand the learned features that can guide the design of efficient ionic molecules. In this work, we develop both fingerprint-based ML models and Graph Neural Networks (GNNs) to predict the CO$_2$ absorption in ILs. Fingerprint works on graph structure at the feature extraction stage, while GNNs directly handle molecule structure in both the feature extraction and model prediction stage. We show that our method outperforms previous ML models by reaching a high accuracy (MAE of 0.0137, $R^2$ of 0.9884). Furthermore, we take the advantage of GNNs feature representation and develop a substructure-based explanation method that provides insight into how each chemical fragments within IL molecules contribute to the CO$_2$ absorption prediction of ML models. We also show that our explanation result agrees with some ground truth from the theoretical reaction mechanism of CO$_2$ absorption in ILs, which can advise on the design of novel and efficient functional ILs in the future.