 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLess is more: sampling chemical space with active learning
Apr 09, 2018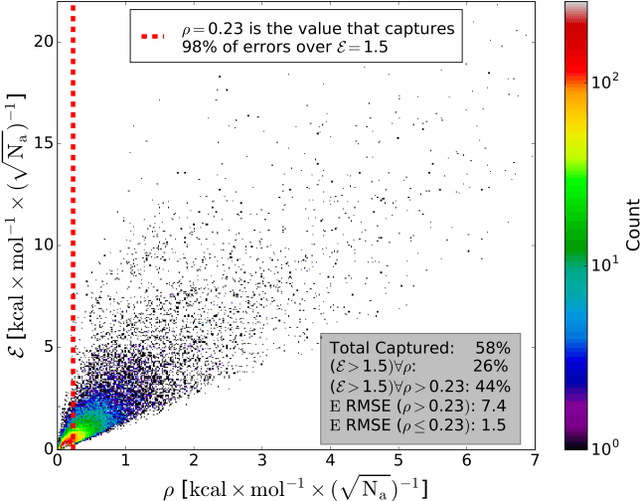
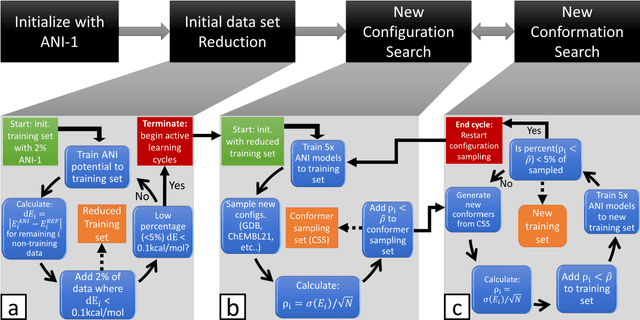
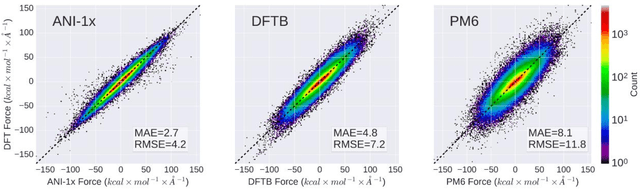
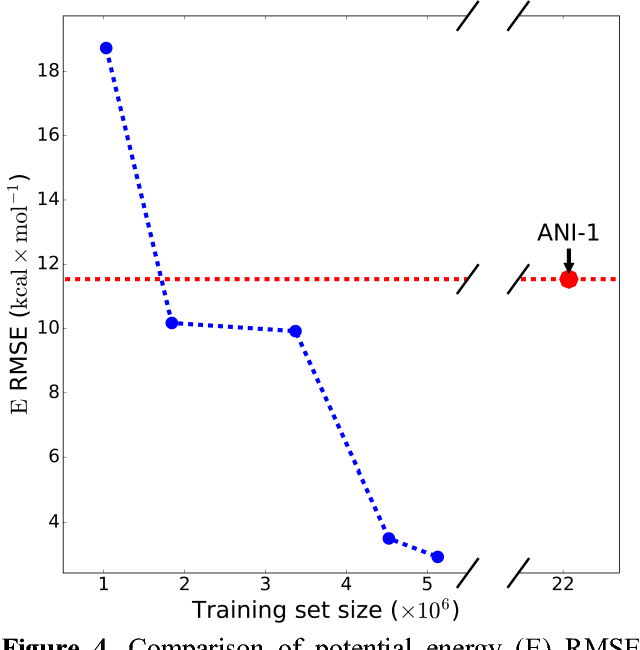
The development of accurate and transferable machine learning (ML) potentials for predicting molecular energetics is a challenging task. The process of data generation to train such ML potentials is a task neither well understood nor researched in detail. In this work, we present a fully automated approach for the generation of datasets with the intent of training universal ML potentials. It is based on the concept of active learning (AL) via Query by Committee (QBC), which uses the disagreement between an ensemble of ML potentials to infer the reliability of the ensemble's prediction. QBC allows the presented AL algorithm to automatically sample regions of chemical space where the ML potential fails to accurately predict the potential energy. AL improves the overall fitness of ANAKIN-ME (ANI) deep learning potentials in rigorous test cases by mitigating human biases in deciding what new training data to use. AL also reduces the training set size to a fraction of the data required when using naive random sampling techniques. To provide validation of our AL approach we develop the COMP6 benchmark (publicly available on GitHub), which contains a diverse set of organic molecules. Through the AL process, it is shown that the AL-based potentials perform as well as the ANI-1 potential on COMP6 with only 10% of the data, and vastly outperforms ANI-1 with 25% the amount of data. Finally, we show that our proposed AL technique develops a universal ANI potential (ANI-1x) that provides accurate energy and force predictions on the entire COMP6 benchmark. This universal ML potential achieves a level of accuracy on par with the best ML potentials for single molecule or materials, while remaining applicable to the general class of organic molecules comprised of the elements CHNO.
* Accepted at J. Chem. Phys
ANI-1: A data set of 20M off-equilibrium DFT calculations for organic molecules
Dec 12, 2017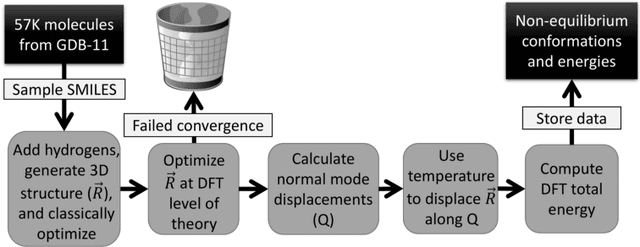
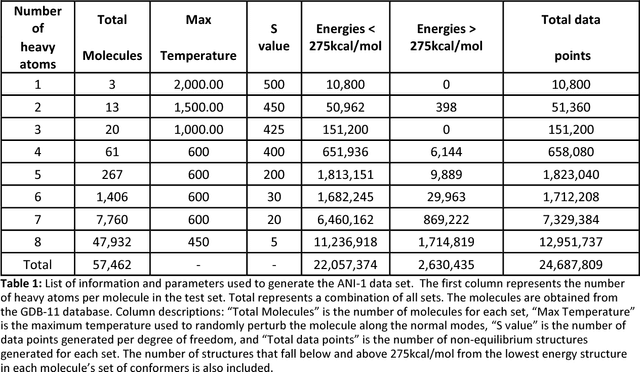
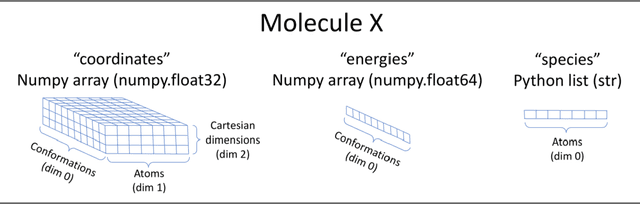
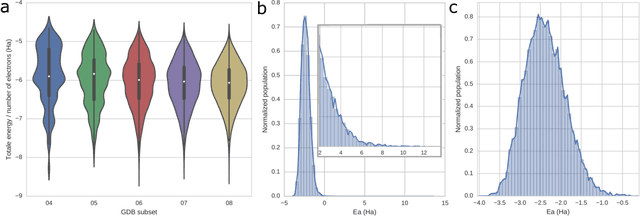
One of the grand challenges in modern theoretical chemistry is designing and implementing approximations that expedite ab initio methods without loss of accuracy. Machine learning (ML), in particular neural networks, are emerging as a powerful approach to constructing various forms of transferable atomistic potentials. They have been successfully applied in a variety of applications in chemistry, biology, catalysis, and solid-state physics. However, these models are heavily dependent on the quality and quantity of data used in their fitting. Fitting highly flexible ML potentials comes at a cost: a vast amount of reference data is required to properly train these models. We address this need by providing access to a large computational DFT database, which consists of 20M conformations for 57,454 small organic molecules. We believe it will become a new standard benchmark for comparison of current and future methods in the ML potential community.