 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProjected Hessian Learning: Fast Curvature Supervision for Accurate Machine-Learning Interatomic Potentials
Mar 04, 2026The Hessian matrix (second derivatives) encodes far richer local curvature of the potential energy surface than energies and forces alone. However, training machine-learning interatomic potentials (MLIPs) with full Hessians is often impractical because explicitly forming and storing Hessian matrices scales quadratically in cost and memory. We introduce Projected Hessian Learning (PHL), a scalable second-order training framework that injects curvature information using only Hessian-vector products (HVPs). Rather than constructing the Hessian, PHL projects curvature along stochastic probe directions and uses an unbiased stochastic trace-based loss with favorable system-size scaling, enabling curvature-informed training without quadratic memory growth. We benchmark PHL on a chemically diverse dataset of reactants, products, transition states, intrinsic reaction coordinates, and normal-mode sampled geometries computed at omegaB97XD/6-31G(d). We compare energy-force training (E-F), two HVP-based schemes (E-F-HVP with one-hot or randomized probes), and full energy-force-Hessian training (E-F-H). With randomized probes per minibatch, both HVP schemes match full-Hessian training in energy, force, and Hessian accuracy while delivering >24x epoch speedups for the small molecular systems studied. In a fixed-probe regime with one HVP per molecule, randomized projections consistently outperform one-column probing, especially for far-from-equilibrium geometries. Overall, PHL replaces explicit Hessian supervision with force-complexity curvature training, retaining most second-order accuracy gains while scaling to larger, more complex molecular systems.
Discrete Spatial Diffusion: Intensity-Preserving Diffusion Modeling
May 03, 2025Generative diffusion models have achieved remarkable success in producing high-quality images. However, because these models typically operate in continuous intensity spaces - diffusing independently per pixel and color channel - they are fundamentally ill-suited for applications where quantities such as particle counts or material units are inherently discrete and governed by strict conservation laws such as mass preservation, limiting their applicability in scientific workflows. To address this limitation, we propose Discrete Spatial Diffusion (DSD), a framework based on a continuous-time, discrete-state jump stochastic process that operates directly in discrete spatial domains while strictly preserving mass in both forward and reverse diffusion processes. By using spatial diffusion to achieve mass preservation, we introduce stochasticity naturally through a discrete formulation. We demonstrate the expressive flexibility of DSD by performing image synthesis, class conditioning, and image inpainting across widely-used image benchmarks, with the ability to condition on image intensity. Additionally, we highlight its applicability to domain-specific scientific data for materials microstructure, bridging the gap between diffusion models and mass-conditioned scientific applications.
Optimal Invariant Bases for Atomistic Machine Learning
Mar 30, 2025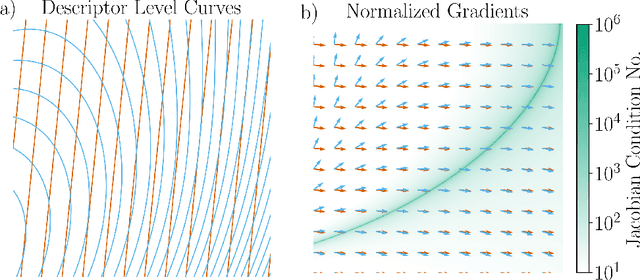
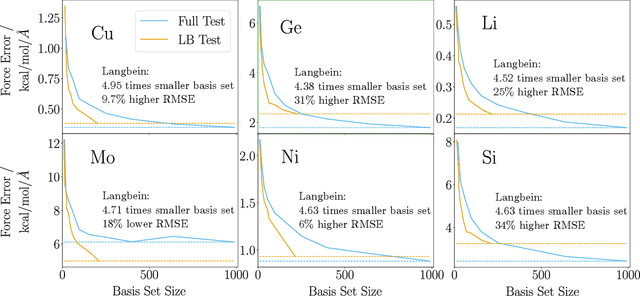
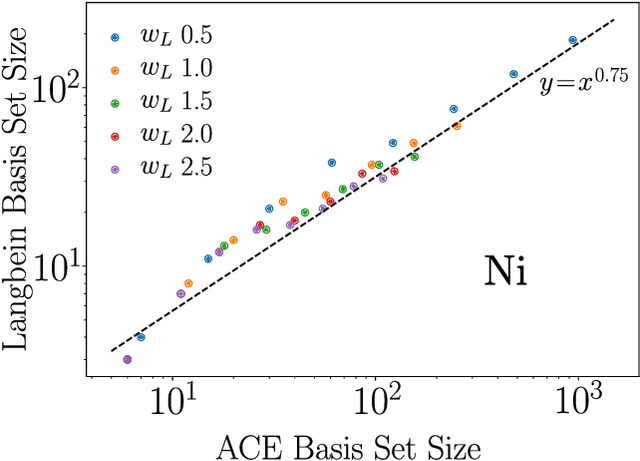
The representation of atomic configurations for machine learning models has led to the development of numerous descriptors, often to describe the local environment of atoms. However, many of these representations are incomplete and/or functionally dependent. Incomplete descriptor sets are unable to represent all meaningful changes in the atomic environment. Complete constructions of atomic environment descriptors, on the other hand, often suffer from a high degree of functional dependence, where some descriptors can be written as functions of the others. These redundant descriptors do not provide additional power to discriminate between different atomic environments and increase the computational burden. By employing techniques from the pattern recognition literature to existing atomistic representations, we remove descriptors that are functions of other descriptors to produce the smallest possible set that satisfies completeness. We apply this in two ways: first we refine an existing description, the Atomistic Cluster Expansion. We show that this yields a more efficient subset of descriptors. Second, we augment an incomplete construction based on a scalar neural network, yielding a new message-passing network architecture that can recognize up to 5-body patterns in each neuron by taking advantage of an optimal set of Cartesian tensor invariants. This architecture shows strong accuracy on state-of-the-art benchmarks while retaining low computational cost. Our results not only yield improved models, but point the way to classes of invariant bases that minimize cost while maximizing expressivity for a host of applications.
Flexible Moment-Invariant Bases from Irreducible Tensors
Mar 27, 2025Moment invariants are a powerful tool for the generation of rotation-invariant descriptors needed for many applications in pattern detection, classification, and machine learning. A set of invariants is optimal if it is complete, independent, and robust against degeneracy in the input. In this paper, we show that the current state of the art for the generation of these bases of moment invariants, despite being robust against moment tensors being identically zero, is vulnerable to a degeneracy that is common in real-world applications, namely spherical functions. We show how to overcome this vulnerability by combining two popular moment invariant approaches: one based on spherical harmonics and one based on Cartesian tensor algebra.
Ensemble Knowledge Distillation for Machine Learning Interatomic Potentials
Mar 19, 2025Machine learning interatomic potentials (MLIPs) are a promising tool to accelerate atomistic simulations and molecular property prediction. The quality of MLIPs strongly depends on the quantity of available training data as well as the quantum chemistry (QC) level of theory used to generate that data. Datasets generated with high-fidelity QC methods, such as coupled cluster, are typically restricted to small molecules and may be missing energy gradients. With this limited quantity of data, it is often difficult to train good MLIP models. We present an ensemble knowledge distillation (EKD) method to improve MLIP accuracy when trained to energy-only datasets. In our EKD approach, first, multiple teacher models are trained to QC energies and then used to generate atomic forces for all configurations in the dataset. Next, a student MLIP is trained to both QC energies and to ensemble-averaged forces generated by the teacher models. We apply this workflow on the ANI-1ccx dataset which consists of organic molecules with configuration energies computed at the coupled cluster level of theory. The resulting student MLIPs achieve new state-of-the-art accuracy on the out-of-sample COMP6 benchmark and improved stability for molecular dynamics simulations. The EKD approach for MLIP is broadly applicable for chemical, biomolecular and materials science simulations.
Model-Agnostic Knowledge Guided Correction for Improved Neural Surrogate Rollout
Mar 13, 2025Modeling the evolution of physical systems is critical to many applications in science and engineering. As the evolution of these systems is governed by partial differential equations (PDEs), there are a number of computational simulations which resolve these systems with high accuracy. However, as these simulations incur high computational costs, they are infeasible to be employed for large-scale analysis. A popular alternative to simulators are neural network surrogates which are trained in a data-driven manner and are much more computationally efficient. However, these surrogate models suffer from high rollout error when used autoregressively, especially when confronted with training data paucity. Existing work proposes to improve surrogate rollout error by either including physical loss terms directly in the optimization of the model or incorporating computational simulators as `differentiable layers' in the neural network. Both of these approaches have their challenges, with physical loss functions suffering from slow convergence for stiff PDEs and simulator layers requiring gradients which are not always available, especially in legacy simulators. We propose the Hybrid PDE Predictor with Reinforcement Learning (HyPER) model: a model-agnostic, RL based, cost-aware model which combines a neural surrogate, RL decision model, and a physics simulator (with or without gradients) to reduce surrogate rollout error significantly. In addition to reducing in-distribution rollout error by **47%-78%**, HyPER learns an intelligent policy that is adaptable to changing physical conditions and resistant to noise corruption. Code available at https://github.com/scailab/HyPER.
Teacher-student training improves accuracy and efficiency of machine learning inter-atomic potentials
Feb 07, 2025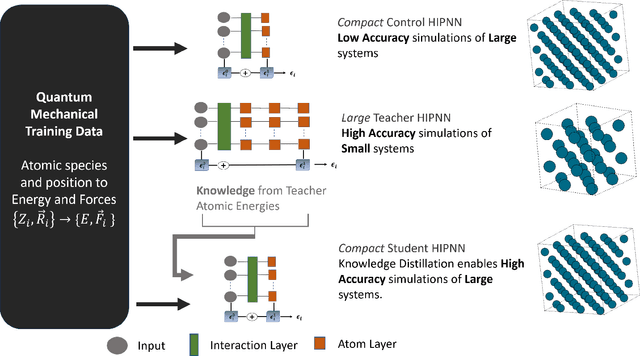

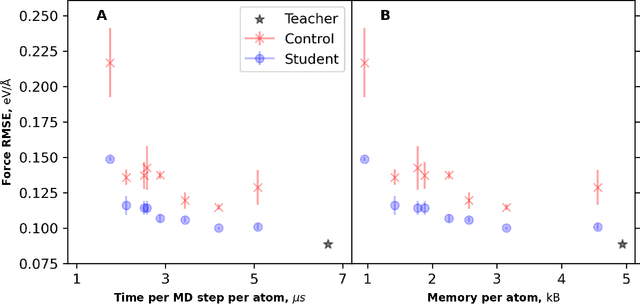
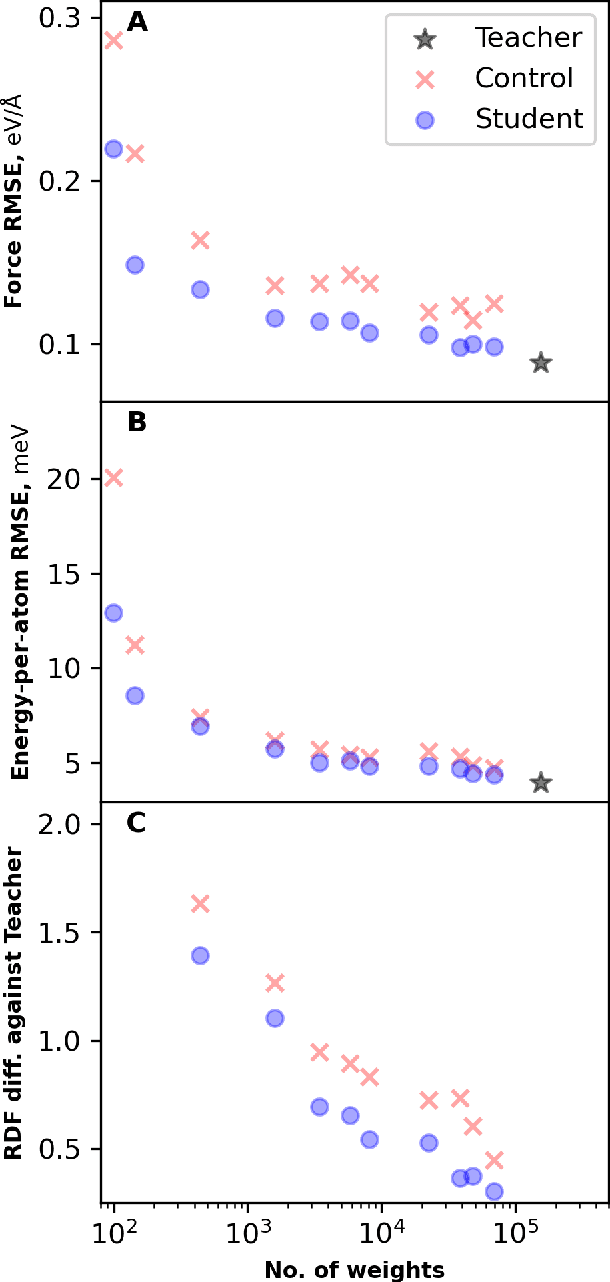
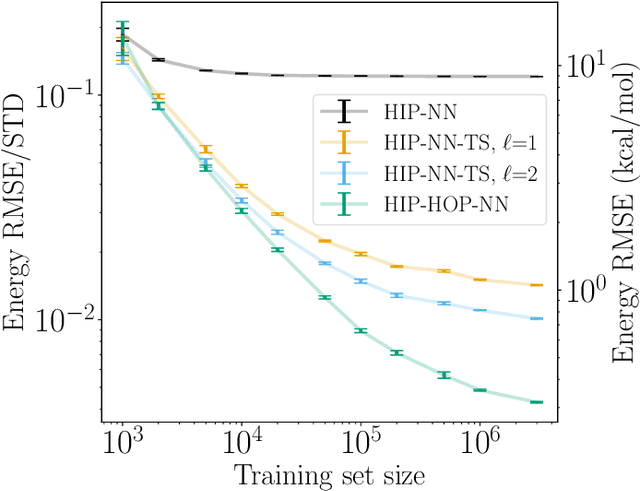
Machine learning inter-atomic potentials (MLIPs) are revolutionizing the field of molecular dynamics (MD) simulations. Recent MLIPs have tended towards more complex architectures trained on larger datasets. The resulting increase in computational and memory costs may prohibit the application of these MLIPs to perform large-scale MD simulations. Here, we present a teacher-student training framework in which the latent knowledge from the teacher (atomic energies) is used to augment the students' training. We show that the light-weight student MLIPs have faster MD speeds at a fraction of the memory footprint compared to the teacher models. Remarkably, the student models can even surpass the accuracy of the teachers, even though both are trained on the same quantum chemistry dataset. Our work highlights a practical method for MLIPs to reduce the resources required for large-scale MD simulations.
Thermodynamic Transferability in Coarse-Grained Force Fields using Graph Neural Networks
Jun 17, 2024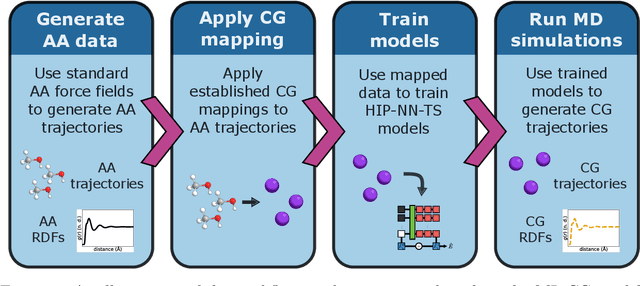
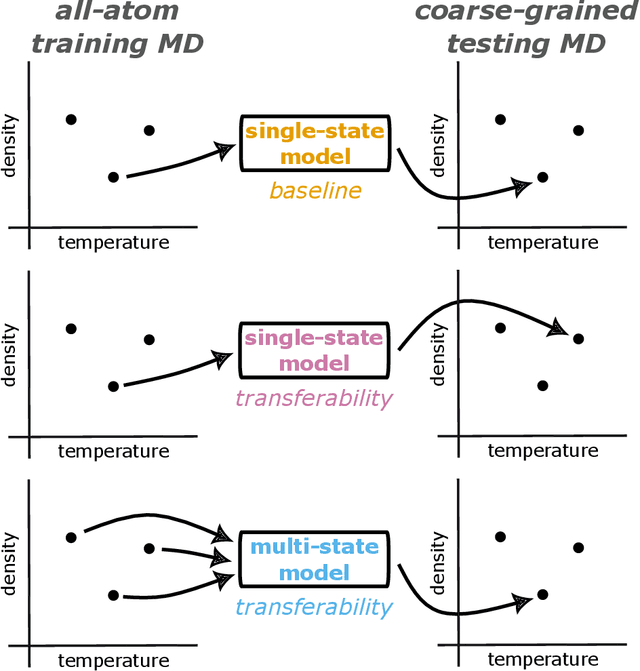
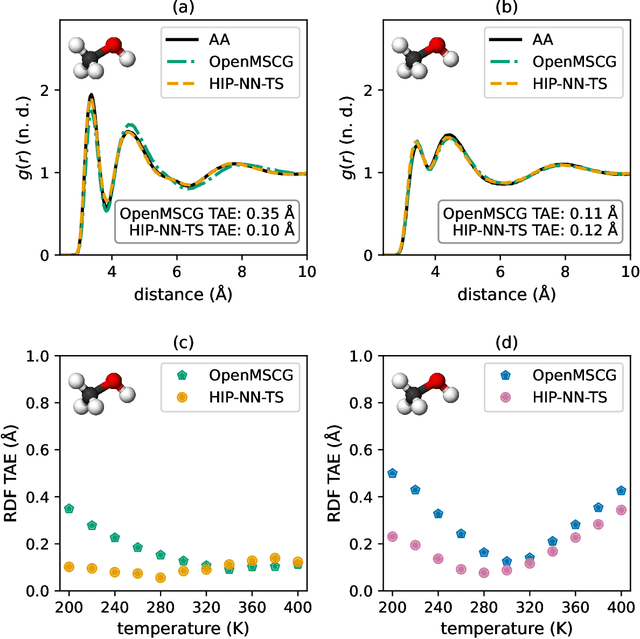
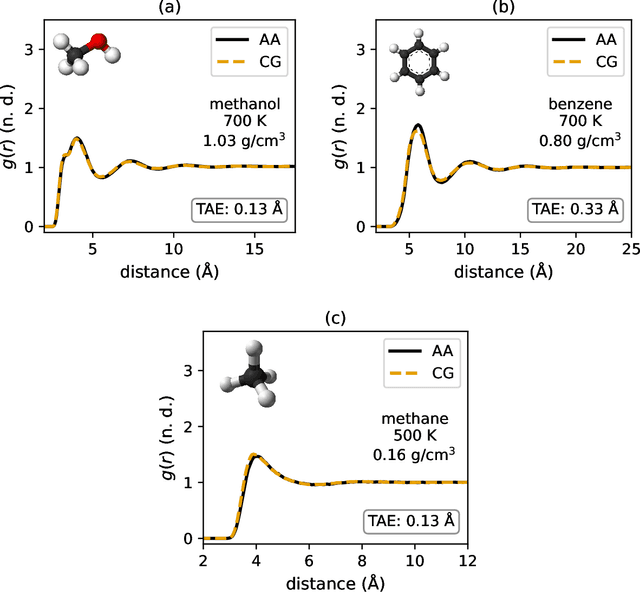
Coarse-graining is a molecular modeling technique in which an atomistic system is represented in a simplified fashion that retains the most significant system features that contribute to a target output, while removing the degrees of freedom that are less relevant. This reduction in model complexity allows coarse-grained molecular simulations to reach increased spatial and temporal scales compared to corresponding all-atom models. A core challenge in coarse-graining is to construct a force field that represents the interactions in the new representation in a way that preserves the atomistic-level properties. Many approaches to building coarse-grained force fields have limited transferability between different thermodynamic conditions as a result of averaging over internal fluctuations at a specific thermodynamic state point. Here, we use a graph-convolutional neural network architecture, the Hierarchically Interacting Particle Neural Network with Tensor Sensitivity (HIP-NN-TS), to develop a highly automated training pipeline for coarse grained force fields which allows for studying the transferability of coarse-grained models based on the force-matching approach. We show that this approach not only yields highly accurate force fields, but also that these force fields are more transferable through a variety of thermodynamic conditions. These results illustrate the potential of machine learning techniques such as graph neural networks to improve the construction of transferable coarse-grained force fields.
Blackout Diffusion: Generative Diffusion Models in Discrete-State Spaces
May 18, 2023



Typical generative diffusion models rely on a Gaussian diffusion process for training the backward transformations, which can then be used to generate samples from Gaussian noise. However, real world data often takes place in discrete-state spaces, including many scientific applications. Here, we develop a theoretical formulation for arbitrary discrete-state Markov processes in the forward diffusion process using exact (as opposed to variational) analysis. We relate the theory to the existing continuous-state Gaussian diffusion as well as other approaches to discrete diffusion, and identify the corresponding reverse-time stochastic process and score function in the continuous-time setting, and the reverse-time mapping in the discrete-time setting. As an example of this framework, we introduce ``Blackout Diffusion'', which learns to produce samples from an empty image instead of from noise. Numerical experiments on the CIFAR-10, Binarized MNIST, and CelebA datasets confirm the feasibility of our approach. Generalizing from specific (Gaussian) forward processes to discrete-state processes without a variational approximation sheds light on how to interpret diffusion models, which we discuss.
Predictive Scale-Bridging Simulations through Active Learning
Sep 20, 2022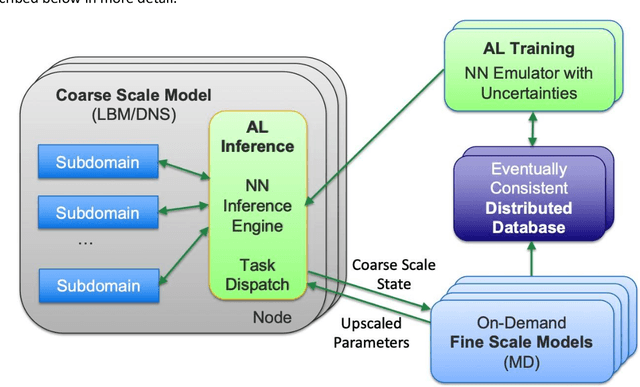
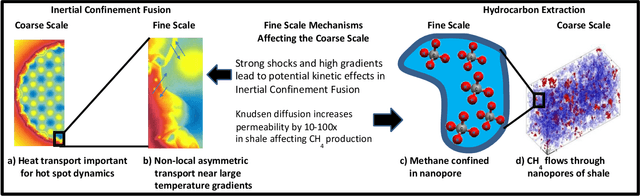
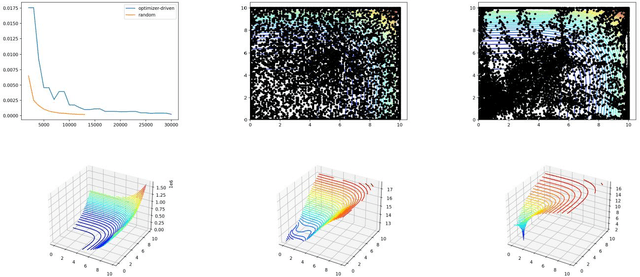
Throughout computational science, there is a growing need to utilize the continual improvements in raw computational horsepower to achieve greater physical fidelity through scale-bridging over brute-force increases in the number of mesh elements. For instance, quantitative predictions of transport in nanoporous media, critical to hydrocarbon extraction from tight shale formations, are impossible without accounting for molecular-level interactions. Similarly, inertial confinement fusion simulations rely on numerical diffusion to simulate molecular effects such as non-local transport and mixing without truly accounting for molecular interactions. With these two disparate applications in mind, we develop a novel capability which uses an active learning approach to optimize the use of local fine-scale simulations for informing coarse-scale hydrodynamics. Our approach addresses three challenges: forecasting continuum coarse-scale trajectory to speculatively execute new fine-scale molecular dynamics calculations, dynamically updating coarse-scale from fine-scale calculations, and quantifying uncertainty in neural network models.