 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTeacher-student training improves accuracy and efficiency of machine learning inter-atomic potentials
Feb 07, 2025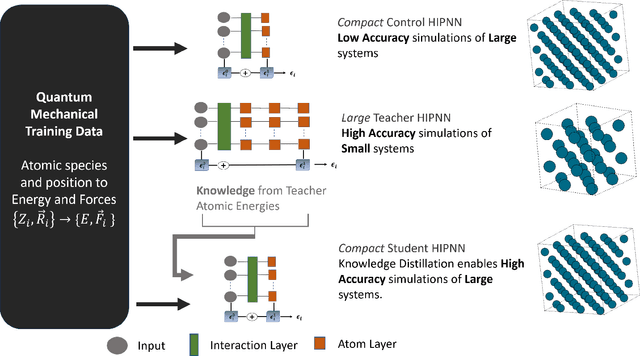

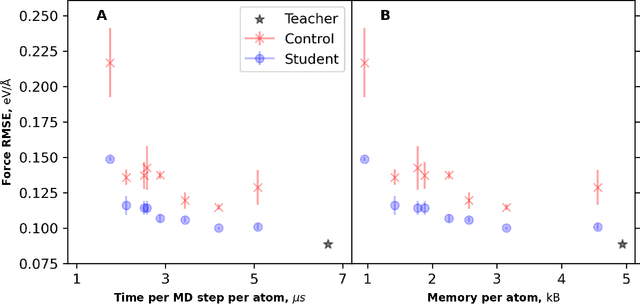
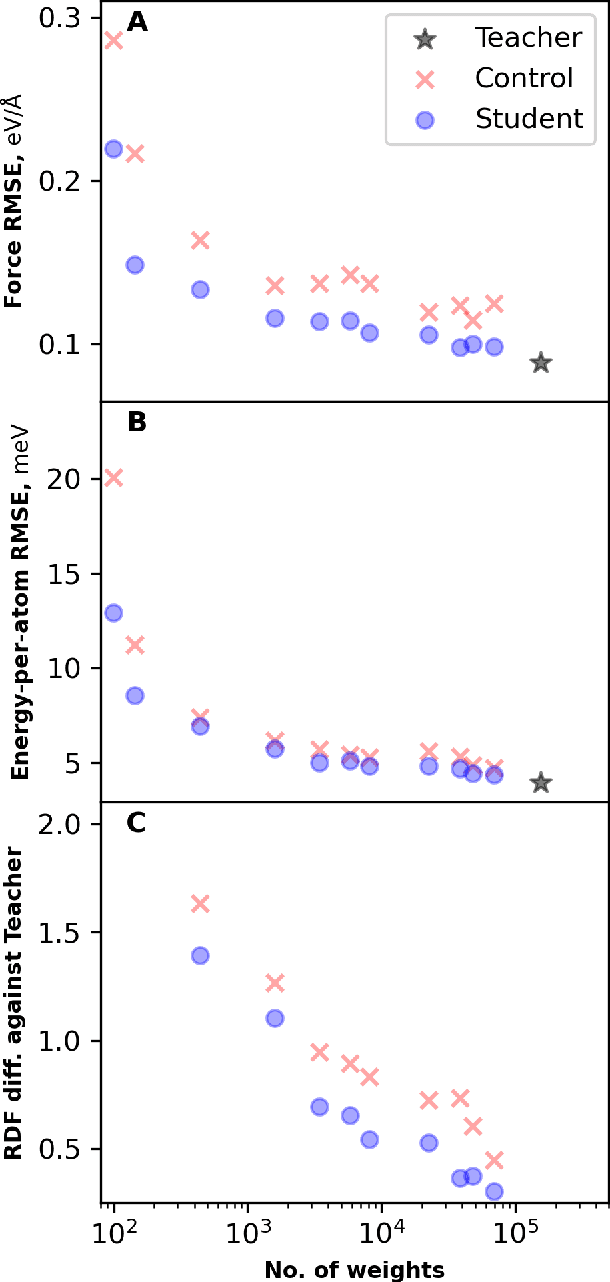
Machine learning inter-atomic potentials (MLIPs) are revolutionizing the field of molecular dynamics (MD) simulations. Recent MLIPs have tended towards more complex architectures trained on larger datasets. The resulting increase in computational and memory costs may prohibit the application of these MLIPs to perform large-scale MD simulations. Here, we present a teacher-student training framework in which the latent knowledge from the teacher (atomic energies) is used to augment the students' training. We show that the light-weight student MLIPs have faster MD speeds at a fraction of the memory footprint compared to the teacher models. Remarkably, the student models can even surpass the accuracy of the teachers, even though both are trained on the same quantum chemistry dataset. Our work highlights a practical method for MLIPs to reduce the resources required for large-scale MD simulations.
Automated discovery of a robust interatomic potential for aluminum
Mar 10, 2020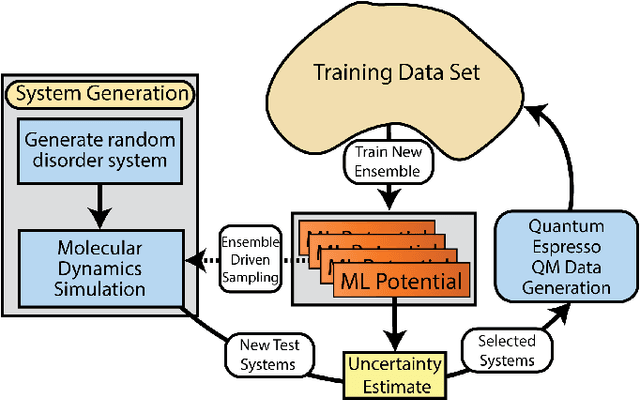
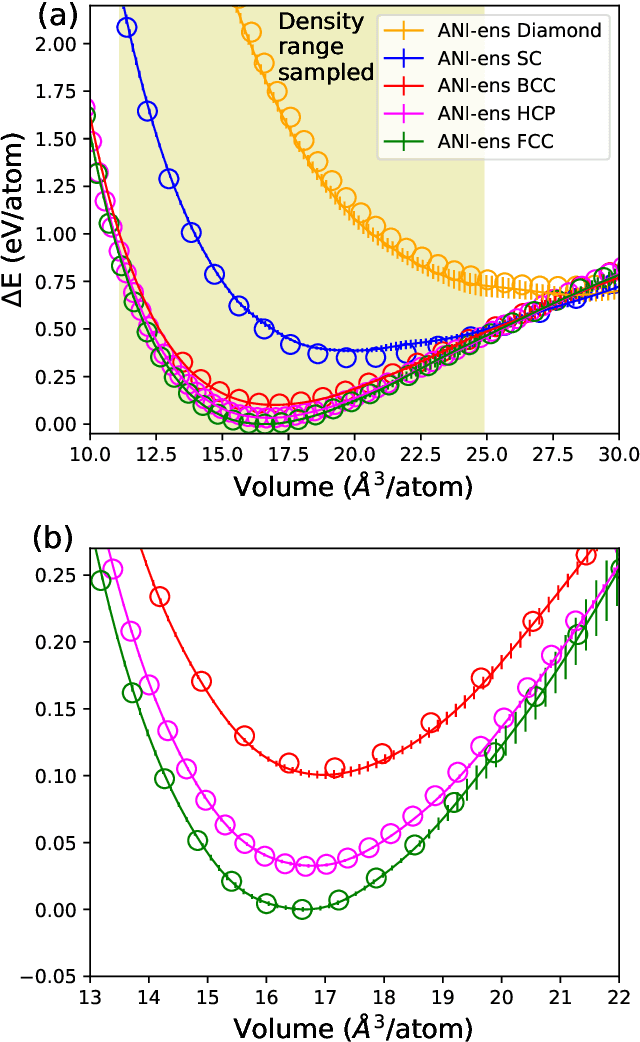
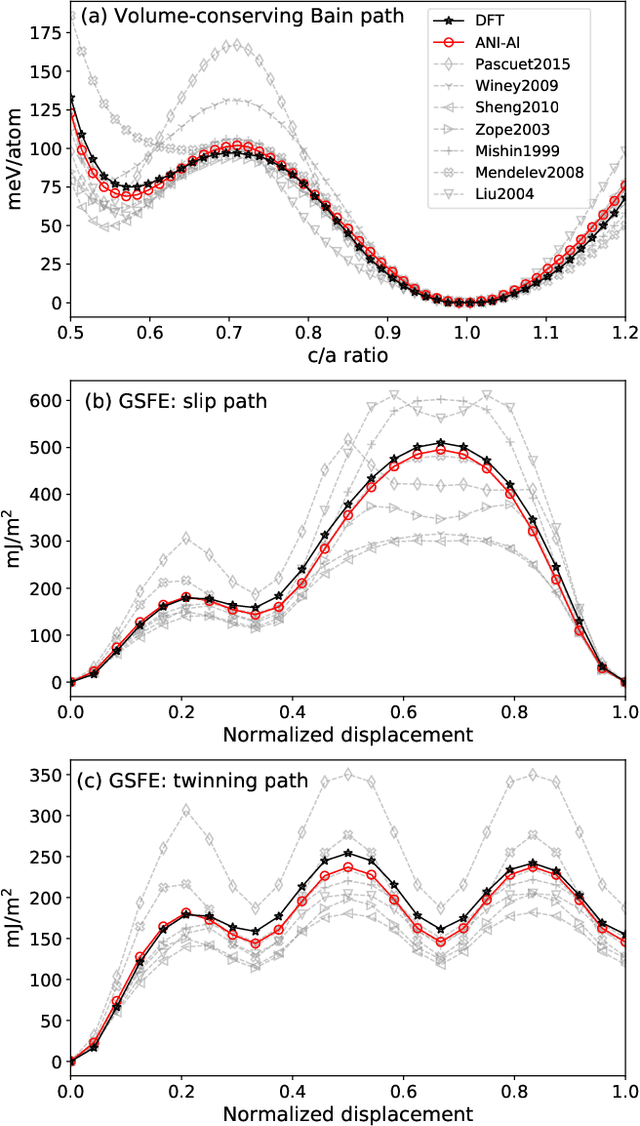
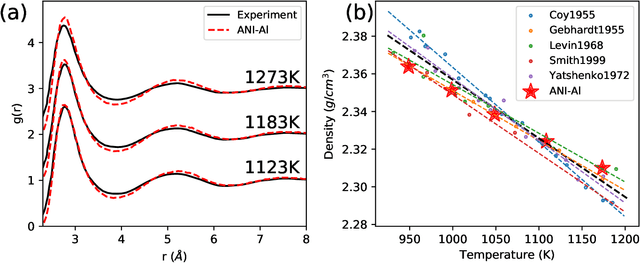
Atomistic molecular dynamics simulation is an important tool for predicting materials properties. Accuracy depends crucially on the model for the interatomic potential. The gold standard would be quantum mechanics (QM) based force calculations, but such a first-principles approach becomes prohibitively expensive at large system sizes. Efficient machine learning models (ML) have become increasingly popular as surrogates for QM. Neural networks with many thousands of parameters excel in capturing structure within a large dataset, but may struggle to extrapolate beyond the scope of the available data. Here we present a highly automated active learning approach to iteratively collect new QM data that best resolves weaknesses in the existing ML model. We exemplify our approach by developing a general potential for elemental aluminum. At each active learning iteration, the method (1) trains an ANI-style neural network potential from the available data, (2) uses this potential to drive molecular dynamics simulations, and (3) collects new QM data whenever the neural network identifies an atomic configuration for which it cannot make a good prediction. All molecular dynamics simulations are initialized to a disordered configuration, and then driven according to randomized, time-varying temperatures. This nonequilibrium molecular dynamics forms a variety of crystalline and defected configurations. By training on all such automatically collected data, we produce ANI-Al, our new interatomic potential for aluminum. We demonstrate the remarkable transferability of ANI-Al by benchmarking against experimental data, e.g., the radial distribution function in melt, various properties of the stable face-centered cubic (FCC) crystal, and the coexistence curve between melt and FCC.