 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDifferentiable multiphase flow model for physics-informed machine learning in reservoir pressure management
Aug 26, 2025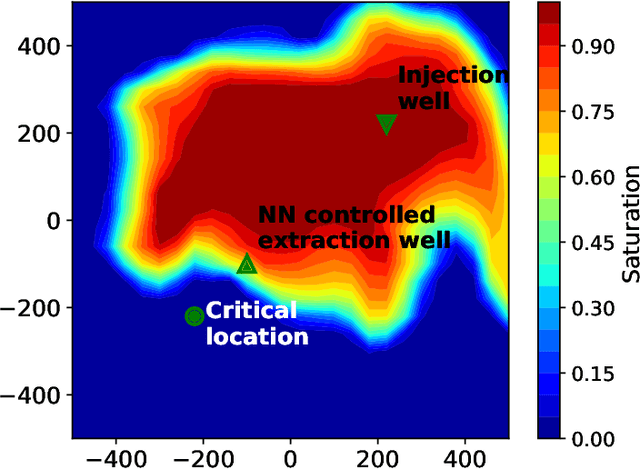
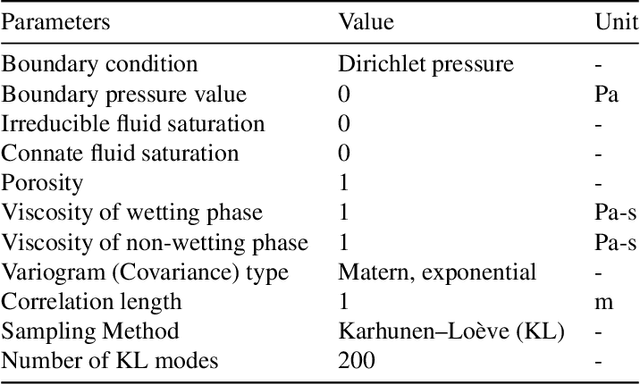
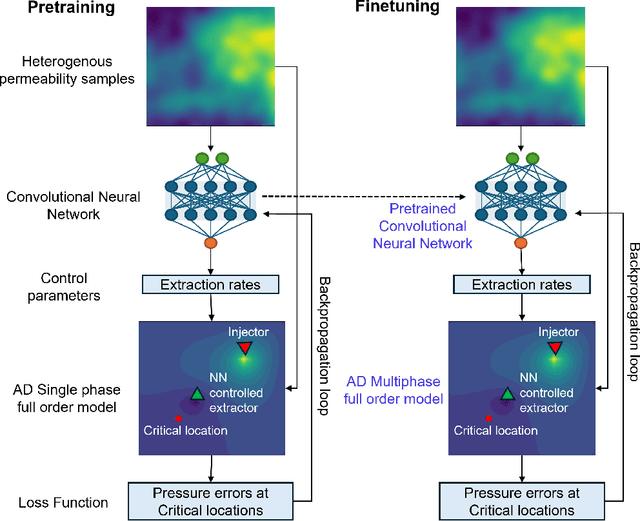
Accurate subsurface reservoir pressure control is extremely challenging due to geological heterogeneity and multiphase fluid-flow dynamics. Predicting behavior in this setting relies on high-fidelity physics-based simulations that are computationally expensive. Yet, the uncertain, heterogeneous properties that control these flows make it necessary to perform many of these expensive simulations, which is often prohibitive. To address these challenges, we introduce a physics-informed machine learning workflow that couples a fully differentiable multiphase flow simulator, which is implemented in the DPFEHM framework with a convolutional neural network (CNN). The CNN learns to predict fluid extraction rates from heterogeneous permeability fields to enforce pressure limits at critical reservoir locations. By incorporating transient multiphase flow physics into the training process, our method enables more practical and accurate predictions for realistic injection-extraction scenarios compare to previous works. To speed up training, we pretrain the model on single-phase, steady-state simulations and then fine-tune it on full multiphase scenarios, which dramatically reduces the computational cost. We demonstrate that high-accuracy training can be achieved with fewer than three thousand full-physics multiphase flow simulations -- compared to previous estimates requiring up to ten million. This drastic reduction in the number of simulations is achieved by leveraging transfer learning from much less expensive single-phase simulations.
Ensemble Knowledge Distillation for Machine Learning Interatomic Potentials
Mar 19, 2025Machine learning interatomic potentials (MLIPs) are a promising tool to accelerate atomistic simulations and molecular property prediction. The quality of MLIPs strongly depends on the quantity of available training data as well as the quantum chemistry (QC) level of theory used to generate that data. Datasets generated with high-fidelity QC methods, such as coupled cluster, are typically restricted to small molecules and may be missing energy gradients. With this limited quantity of data, it is often difficult to train good MLIP models. We present an ensemble knowledge distillation (EKD) method to improve MLIP accuracy when trained to energy-only datasets. In our EKD approach, first, multiple teacher models are trained to QC energies and then used to generate atomic forces for all configurations in the dataset. Next, a student MLIP is trained to both QC energies and to ensemble-averaged forces generated by the teacher models. We apply this workflow on the ANI-1ccx dataset which consists of organic molecules with configuration energies computed at the coupled cluster level of theory. The resulting student MLIPs achieve new state-of-the-art accuracy on the out-of-sample COMP6 benchmark and improved stability for molecular dynamics simulations. The EKD approach for MLIP is broadly applicable for chemical, biomolecular and materials science simulations.
Teacher-student training improves accuracy and efficiency of machine learning inter-atomic potentials
Feb 07, 2025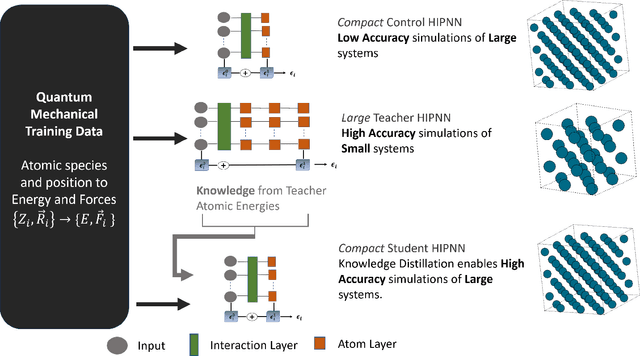

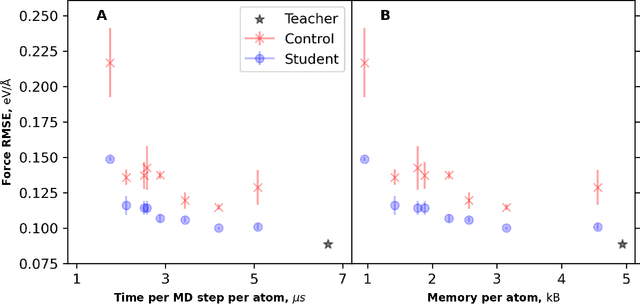
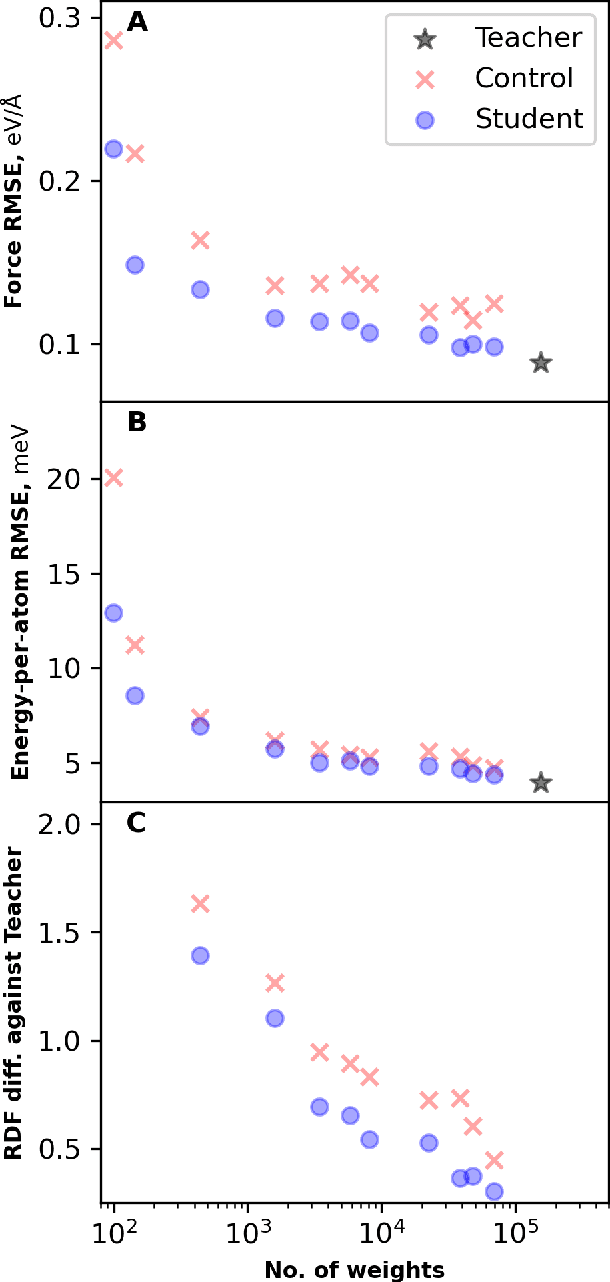
Machine learning inter-atomic potentials (MLIPs) are revolutionizing the field of molecular dynamics (MD) simulations. Recent MLIPs have tended towards more complex architectures trained on larger datasets. The resulting increase in computational and memory costs may prohibit the application of these MLIPs to perform large-scale MD simulations. Here, we present a teacher-student training framework in which the latent knowledge from the teacher (atomic energies) is used to augment the students' training. We show that the light-weight student MLIPs have faster MD speeds at a fraction of the memory footprint compared to the teacher models. Remarkably, the student models can even surpass the accuracy of the teachers, even though both are trained on the same quantum chemistry dataset. Our work highlights a practical method for MLIPs to reduce the resources required for large-scale MD simulations.
Thermodynamic Transferability in Coarse-Grained Force Fields using Graph Neural Networks
Jun 17, 2024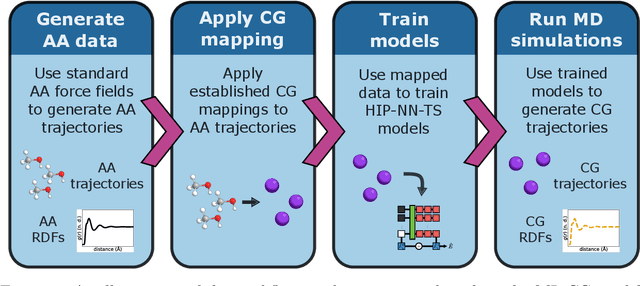
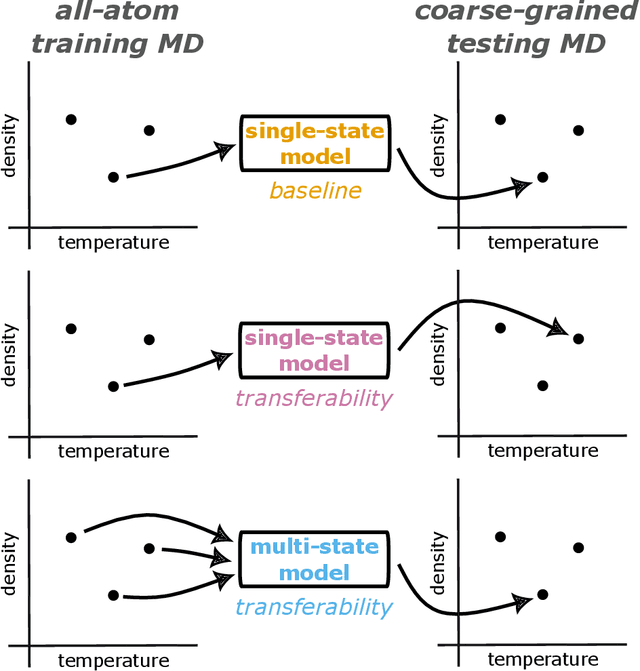
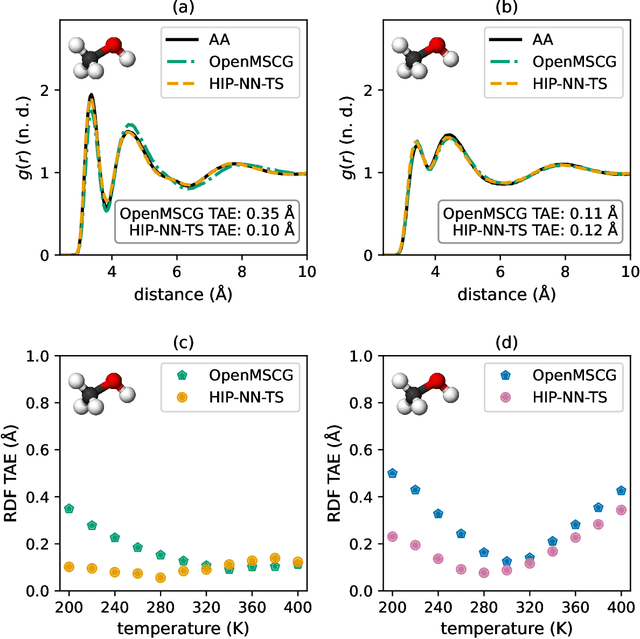
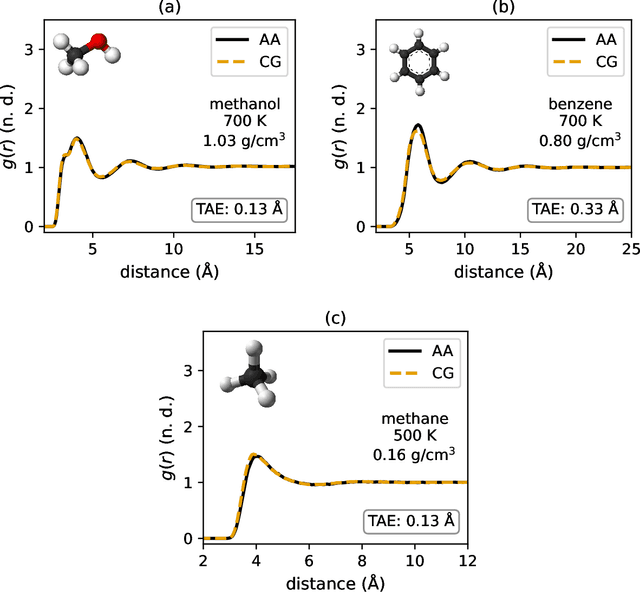
Coarse-graining is a molecular modeling technique in which an atomistic system is represented in a simplified fashion that retains the most significant system features that contribute to a target output, while removing the degrees of freedom that are less relevant. This reduction in model complexity allows coarse-grained molecular simulations to reach increased spatial and temporal scales compared to corresponding all-atom models. A core challenge in coarse-graining is to construct a force field that represents the interactions in the new representation in a way that preserves the atomistic-level properties. Many approaches to building coarse-grained force fields have limited transferability between different thermodynamic conditions as a result of averaging over internal fluctuations at a specific thermodynamic state point. Here, we use a graph-convolutional neural network architecture, the Hierarchically Interacting Particle Neural Network with Tensor Sensitivity (HIP-NN-TS), to develop a highly automated training pipeline for coarse grained force fields which allows for studying the transferability of coarse-grained models based on the force-matching approach. We show that this approach not only yields highly accurate force fields, but also that these force fields are more transferable through a variety of thermodynamic conditions. These results illustrate the potential of machine learning techniques such as graph neural networks to improve the construction of transferable coarse-grained force fields.
Learning the Factors Controlling Mineralization for Geologic Carbon Sequestration
Dec 20, 2023We perform a set of flow and reactive transport simulations within three-dimensional fracture networks to learn the factors controlling mineral reactions. CO$_2$ mineralization requires CO$_2$-laden water, dissolution of a mineral that then leads to precipitation of a CO$_2$-bearing mineral. Our discrete fracture networks (DFN) are partially filled with quartz that gradually dissolves until it reaches a quasi-steady state. At the end of the simulation, we measure the quartz remaining in each fracture within the domain. We observe that a small backbone of fracture exists, where the quartz is fully dissolved which leads to increased flow and transport. However, depending on the DFN topology and the rate of dissolution, we observe a large variability of these changes, which indicates an interplay between the fracture network structure and the impact of geochemical dissolution. In this work, we developed a machine learning framework to extract the important features that support mineralization in the form of dissolution. In addition, we use structural and topological features of the fracture network to predict the remaining quartz volume in quasi-steady state conditions. As a first step to characterizing carbon mineralization, we study dissolution with this framework. We studied a variety of reaction and fracture parameters and their impact on the dissolution of quartz in fracture networks. We found that the dissolution reaction rate constant of quartz and the distance to the flowing backbone in the fracture network are the two most important features that control the amount of quartz left in the system. For the first time, we use a combination of a finite-volume reservoir model and graph-based approach to study reactive transport in a complex fracture network to determine the key features that control dissolution.
Predictive Scale-Bridging Simulations through Active Learning
Sep 20, 2022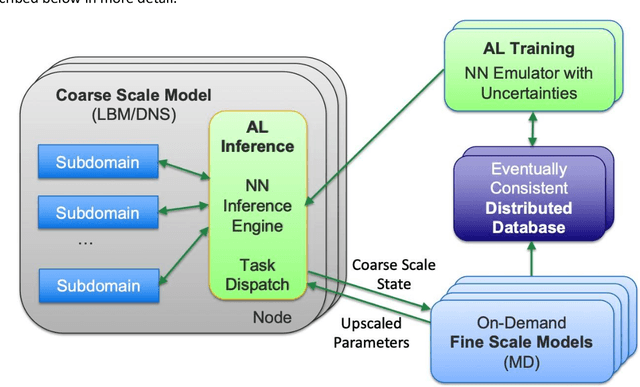
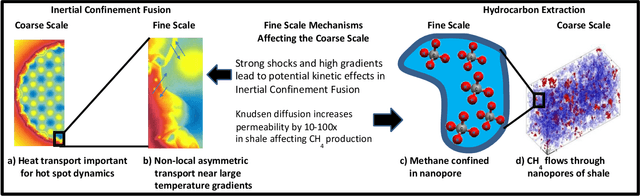
Throughout computational science, there is a growing need to utilize the continual improvements in raw computational horsepower to achieve greater physical fidelity through scale-bridging over brute-force increases in the number of mesh elements. For instance, quantitative predictions of transport in nanoporous media, critical to hydrocarbon extraction from tight shale formations, are impossible without accounting for molecular-level interactions. Similarly, inertial confinement fusion simulations rely on numerical diffusion to simulate molecular effects such as non-local transport and mixing without truly accounting for molecular interactions. With these two disparate applications in mind, we develop a novel capability which uses an active learning approach to optimize the use of local fine-scale simulations for informing coarse-scale hydrodynamics. Our approach addresses three challenges: forecasting continuum coarse-scale trajectory to speculatively execute new fine-scale molecular dynamics calculations, dynamically updating coarse-scale from fine-scale calculations, and quantifying uncertainty in neural network models.
Physics-informed machine learning with differentiable programming for heterogeneous underground reservoir pressure management
Jun 21, 2022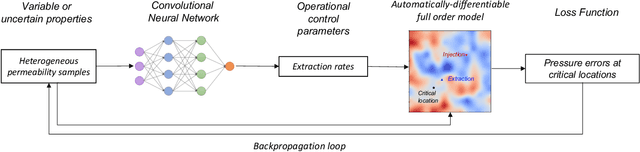

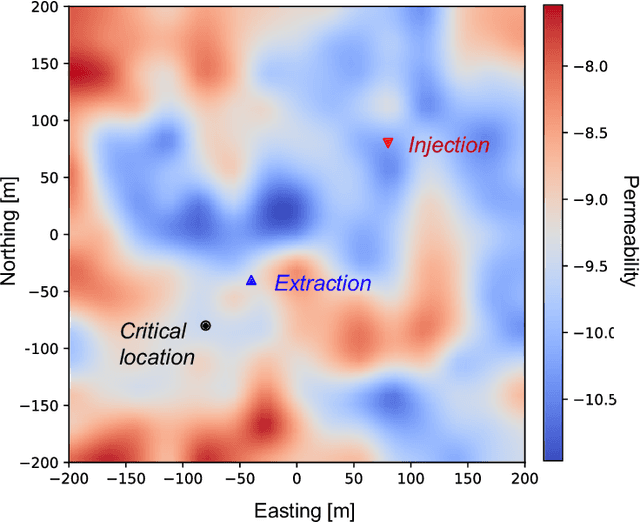
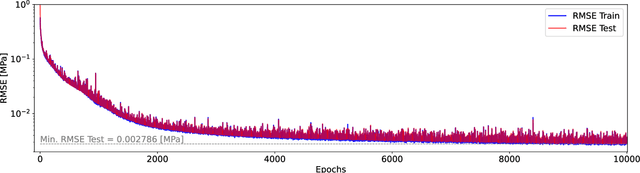
Avoiding over-pressurization in subsurface reservoirs is critical for applications like CO2 sequestration and wastewater injection. Managing the pressures by controlling injection/extraction are challenging because of complex heterogeneity in the subsurface. The heterogeneity typically requires high-fidelity physics-based models to make predictions on CO$_2$ fate. Furthermore, characterizing the heterogeneity accurately is fraught with parametric uncertainty. Accounting for both, heterogeneity and uncertainty, makes this a computationally-intensive problem challenging for current reservoir simulators. To tackle this, we use differentiable programming with a full-physics model and machine learning to determine the fluid extraction rates that prevent over-pressurization at critical reservoir locations. We use DPFEHM framework, which has trustworthy physics based on the standard two-point flux finite volume discretization and is also automatically differentiable like machine learning models. Our physics-informed machine learning framework uses convolutional neural networks to learn an appropriate extraction rate based on the permeability field. We also perform a hyperparameter search to improve the model's accuracy. Training and testing scenarios are executed to evaluate the feasibility of using physics-informed machine learning to manage reservoir pressures. We constructed and tested a sufficiently accurate simulator that is 400000 times faster than the underlying physics-based simulator, allowing for near real-time analysis and robust uncertainty quantification.