 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe impact of conformer quality on learned representations of molecular conformer ensembles
Feb 18, 2025Training machine learning models to predict properties of molecular conformer ensembles is an increasingly popular strategy to accelerate the conformational analysis of drug-like small molecules, reactive organic substrates, and homogeneous catalysts. For high-throughput analyses especially, trained surrogate models can help circumvent traditional approaches to conformational analysis that rely on expensive conformer searches and geometry optimizations. Here, we question how the performance of surrogate models for predicting 3D conformer-dependent properties (of a single, active conformer) is affected by the quality of the 3D conformers used as their input. How well do lower-quality conformers inform the prediction of properties of higher-quality conformers? Does the fidelity of geometry optimization matter when encoding random conformers? For models that encode sets of conformers, how does the presence of the active conformer that induces the target property affect model accuracy? How do predictions from a surrogate model compare to estimating the properties from cheap ensembles themselves? We explore these questions in the context of predicting Sterimol parameters of conformer ensembles optimized with density functional theory. Although answers will be case-specific, our analyses provide a valuable perspective on 3D representation learning models and raise practical considerations regarding when conformer quality matters.
ShEPhERD: Diffusing shape, electrostatics, and pharmacophores for bioisosteric drug design
Oct 22, 2024Engineering molecules to exhibit precise 3D intermolecular interactions with their environment forms the basis of chemical design. In ligand-based drug design, bioisosteric analogues of known bioactive hits are often identified by virtually screening chemical libraries with shape, electrostatic, and pharmacophore similarity scoring functions. We instead hypothesize that a generative model which learns the joint distribution over 3D molecular structures and their interaction profiles may facilitate 3D interaction-aware chemical design. We specifically design ShEPhERD, an SE(3)-equivariant diffusion model which jointly diffuses/denoises 3D molecular graphs and representations of their shapes, electrostatic potential surfaces, and (directional) pharmacophores to/from Gaussian noise. Inspired by traditional ligand discovery, we compose 3D similarity scoring functions to assess ShEPhERD's ability to conditionally generate novel molecules with desired interaction profiles. We demonstrate ShEPhERD's potential for impact via exemplary drug design tasks including natural product ligand hopping, protein-blind bioactive hit diversification, and bioisosteric fragment merging.
Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks
Sep 29, 2023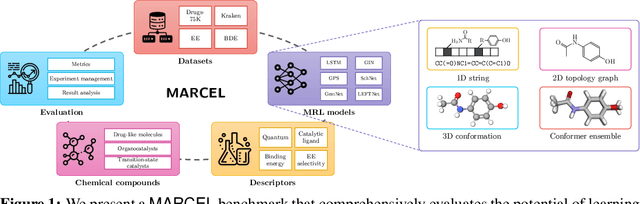
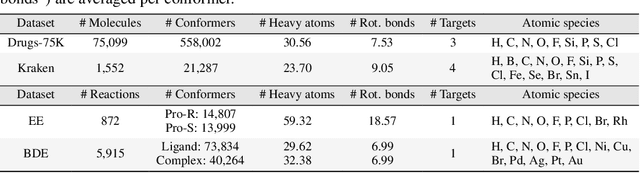
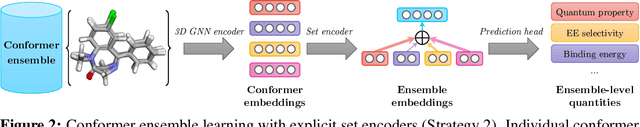
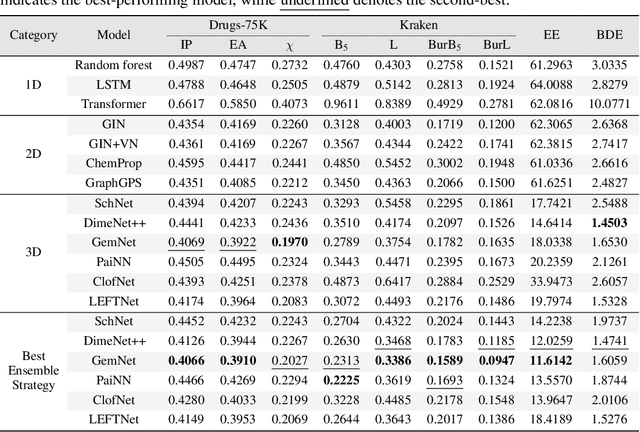
Molecular Representation Learning (MRL) has proven impactful in numerous biochemical applications such as drug discovery and enzyme design. While Graph Neural Networks (GNNs) are effective at learning molecular representations from a 2D molecular graph or a single 3D structure, existing works often overlook the flexible nature of molecules, which continuously interconvert across conformations via chemical bond rotations and minor vibrational perturbations. To better account for molecular flexibility, some recent works formulate MRL as an ensemble learning problem, focusing on explicitly learning from a set of conformer structures. However, most of these studies have limited datasets, tasks, and models. In this work, we introduce the first MoleculAR Conformer Ensemble Learning (MARCEL) benchmark to thoroughly evaluate the potential of learning on conformer ensembles and suggest promising research directions. MARCEL includes four datasets covering diverse molecule- and reaction-level properties of chemically diverse molecules including organocatalysts and transition-metal catalysts, extending beyond the scope of common GNN benchmarks that are confined to drug-like molecules. In addition, we conduct a comprehensive empirical study, which benchmarks representative 1D, 2D, and 3D molecular representation learning models, along with two strategies that explicitly incorporate conformer ensembles into 3D MRL models. Our findings reveal that direct learning from an accessible conformer space can improve performance on a variety of tasks and models.
Artificial Intelligence for Science in Quantum, Atomistic, and Continuum Systems
Jul 17, 2023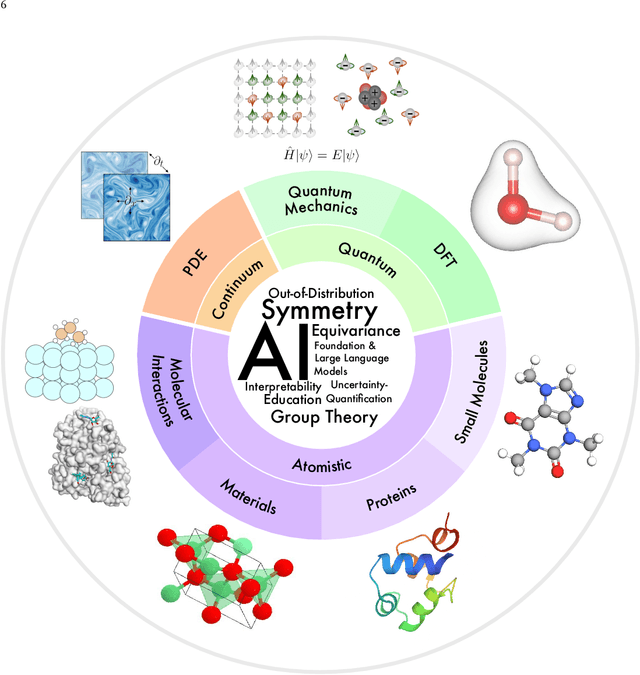
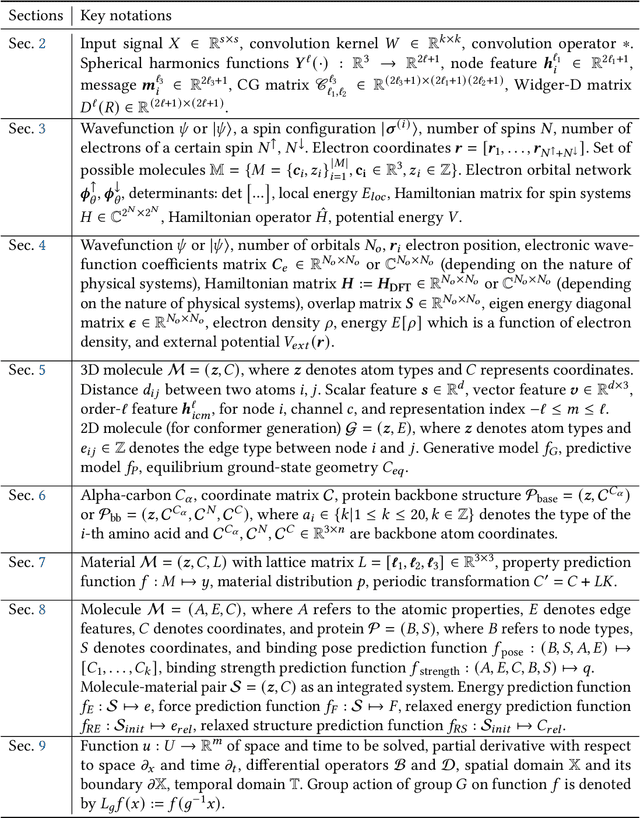
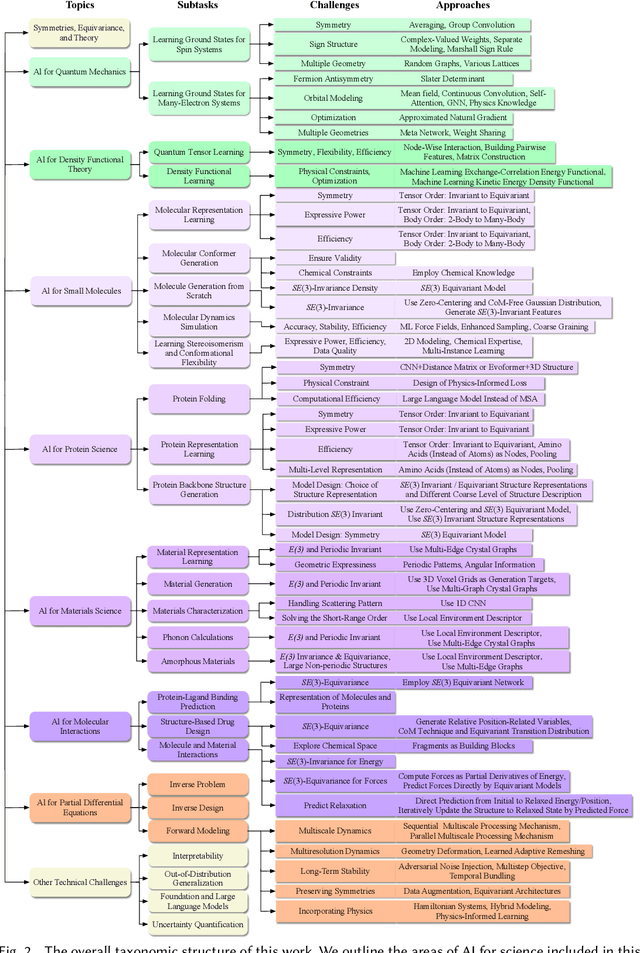

Advances in artificial intelligence (AI) are fueling a new paradigm of discoveries in natural sciences. Today, AI has started to advance natural sciences by improving, accelerating, and enabling our understanding of natural phenomena at a wide range of spatial and temporal scales, giving rise to a new area of research known as AI for science (AI4Science). Being an emerging research paradigm, AI4Science is unique in that it is an enormous and highly interdisciplinary area. Thus, a unified and technical treatment of this field is needed yet challenging. This paper aims to provide a technically thorough account of a subarea of AI4Science; namely, AI for quantum, atomistic, and continuum systems. These areas aim at understanding the physical world from the subatomic (wavefunctions and electron density), atomic (molecules, proteins, materials, and interactions), to macro (fluids, climate, and subsurface) scales and form an important subarea of AI4Science. A unique advantage of focusing on these areas is that they largely share a common set of challenges, thereby allowing a unified and foundational treatment. A key common challenge is how to capture physics first principles, especially symmetries, in natural systems by deep learning methods. We provide an in-depth yet intuitive account of techniques to achieve equivariance to symmetry transformations. We also discuss other common technical challenges, including explainability, out-of-distribution generalization, knowledge transfer with foundation and large language models, and uncertainty quantification. To facilitate learning and education, we provide categorized lists of resources that we found to be useful. We strive to be thorough and unified and hope this initial effort may trigger more community interests and efforts to further advance AI4Science.
Equivariant Shape-Conditioned Generation of 3D Molecules for Ligand-Based Drug Design
Oct 06, 2022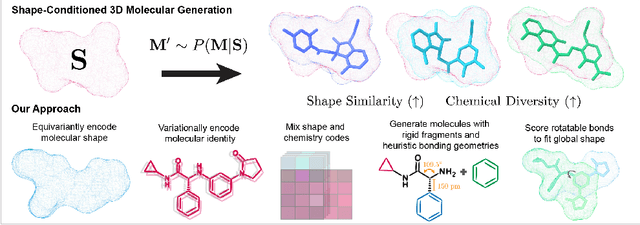
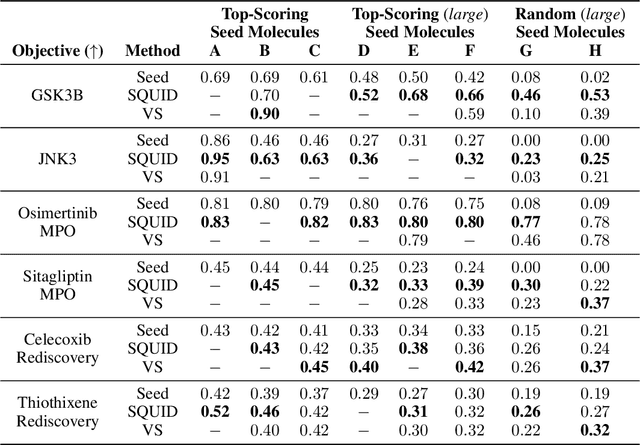
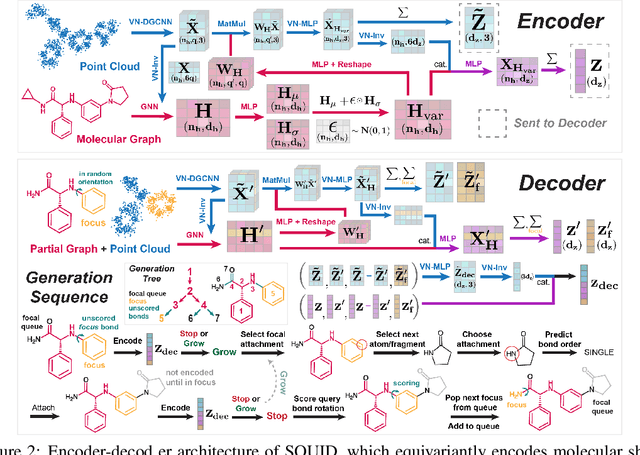

Shape-based virtual screening is widely employed in ligand-based drug design to search chemical libraries for molecules with similar 3D shapes yet novel 2D chemical structures compared to known ligands. 3D deep generative models have the potential to automate this exploration of shape-conditioned 3D chemical space; however, no existing models can reliably generate valid drug-like molecules in conformations that adopt a specific shape such as a known binding pose. We introduce a new multimodal 3D generative model that enables shape-conditioned 3D molecular design by equivariantly encoding molecular shape and variationally encoding chemical identity. We ensure local geometric and chemical validity of generated molecules by using autoregressive fragment-based generation with heuristic bonding geometries, allowing the model to prioritize the scoring of rotatable bonds to best align the growing conformational structure to the target shape. We evaluate our 3D generative model in tasks relevant to drug design including shape-conditioned generation of chemically diverse molecular structures and shape-constrained molecular property optimization, demonstrating its utility over virtual screening of enumerated libraries.
Learning 3D Representations of Molecular Chirality with Invariance to Bond Rotations
Oct 08, 2021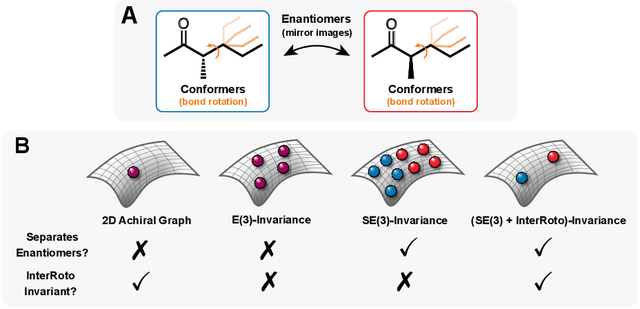

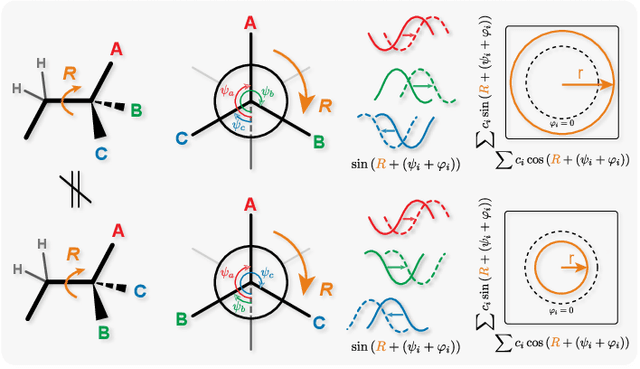
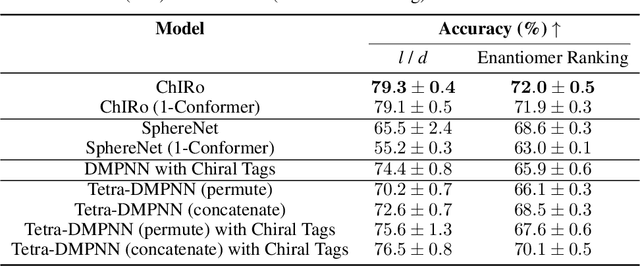
Molecular chirality, a form of stereochemistry most often describing relative spatial arrangements of bonded neighbors around tetrahedral carbon centers, influences the set of 3D conformers accessible to the molecule without changing its 2D graph connectivity. Chirality can strongly alter (bio)chemical interactions, particularly protein-drug binding. Most 2D graph neural networks (GNNs) designed for molecular property prediction at best use atomic labels to na\"ively treat chirality, while E(3)-invariant 3D GNNs are invariant to chirality altogether. To enable representation learning on molecules with defined stereochemistry, we design an SE(3)-invariant model that processes torsion angles of a 3D molecular conformer. We explicitly model conformational flexibility by integrating a novel type of invariance to rotations about internal molecular bonds into the architecture, mitigating the need for multi-conformer data augmentation. We test our model on four benchmarks: contrastive learning to distinguish conformers of different stereoisomers in a learned latent space, classification of chiral centers as R/S, prediction of how enantiomers rotate circularly polarized light, and ranking enantiomers by their docking scores in an enantiosensitive protein pocket. We compare our model, Chiral InterRoto-Invariant Neural Network (ChIRo), with 2D and 3D GNNs to demonstrate that our model achieves state of the art performance when learning chiral-sensitive functions from molecular structures.