 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEquiBind: Geometric Deep Learning for Drug Binding Structure Prediction
Feb 07, 2022
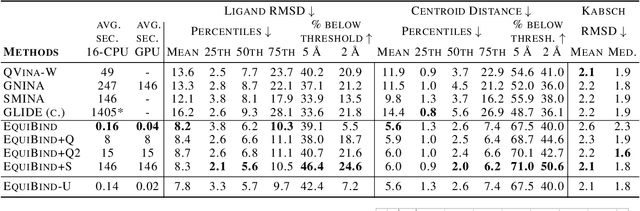

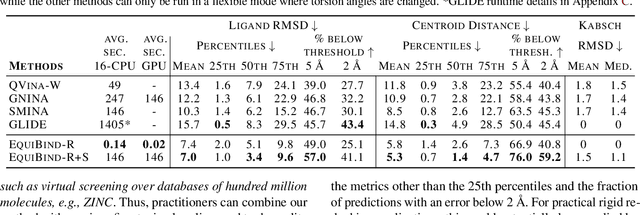
Predicting how a drug-like molecule binds to a specific protein target is a core problem in drug discovery. An extremely fast computational binding method would enable key applications such as fast virtual screening or drug engineering. Existing methods are computationally expensive as they rely on heavy candidate sampling coupled with scoring, ranking, and fine-tuning steps. We challenge this paradigm with EquiBind, an SE(3)-equivariant geometric deep learning model performing direct-shot prediction of both i) the receptor binding location (blind docking) and ii) the ligand's bound pose and orientation. EquiBind achieves significant speed-ups and better quality compared to traditional and recent baselines. Further, we show extra improvements when coupling it with existing fine-tuning techniques at the cost of increased running time. Finally, we propose a novel and fast fine-tuning model that adjusts torsion angles of a ligand's rotatable bonds based on closed-form global minima of the von Mises angular distance to a given input atomic point cloud, avoiding previous expensive differential evolution strategies for energy minimization.
Learning 3D Representations of Molecular Chirality with Invariance to Bond Rotations
Oct 08, 2021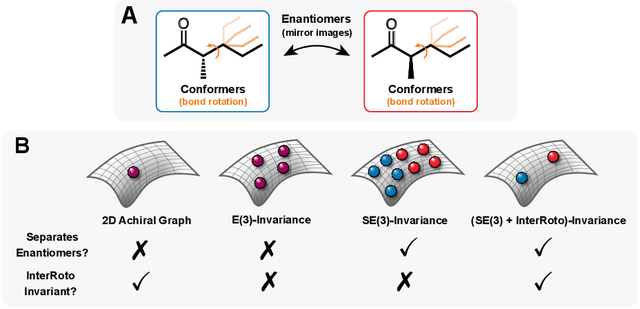

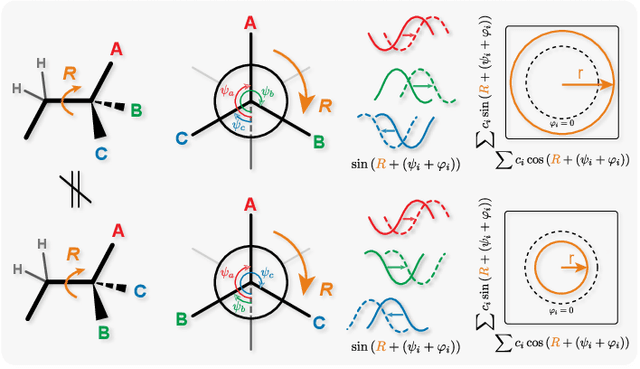
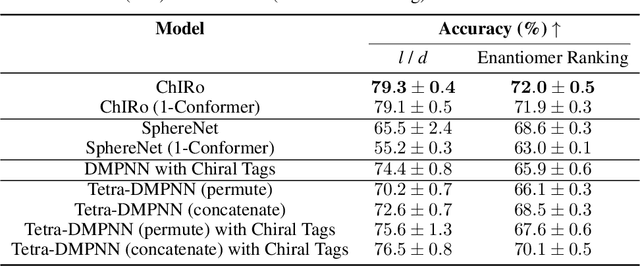
Molecular chirality, a form of stereochemistry most often describing relative spatial arrangements of bonded neighbors around tetrahedral carbon centers, influences the set of 3D conformers accessible to the molecule without changing its 2D graph connectivity. Chirality can strongly alter (bio)chemical interactions, particularly protein-drug binding. Most 2D graph neural networks (GNNs) designed for molecular property prediction at best use atomic labels to na\"ively treat chirality, while E(3)-invariant 3D GNNs are invariant to chirality altogether. To enable representation learning on molecules with defined stereochemistry, we design an SE(3)-invariant model that processes torsion angles of a 3D molecular conformer. We explicitly model conformational flexibility by integrating a novel type of invariance to rotations about internal molecular bonds into the architecture, mitigating the need for multi-conformer data augmentation. We test our model on four benchmarks: contrastive learning to distinguish conformers of different stereoisomers in a learned latent space, classification of chiral centers as R/S, prediction of how enantiomers rotate circularly polarized light, and ranking enantiomers by their docking scores in an enantiosensitive protein pocket. We compare our model, Chiral InterRoto-Invariant Neural Network (ChIRo), with 2D and 3D GNNs to demonstrate that our model achieves state of the art performance when learning chiral-sensitive functions from molecular structures.
GeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles
Jun 08, 2021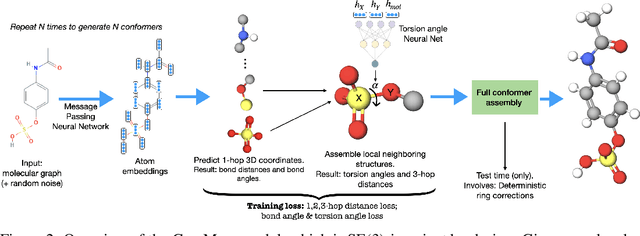
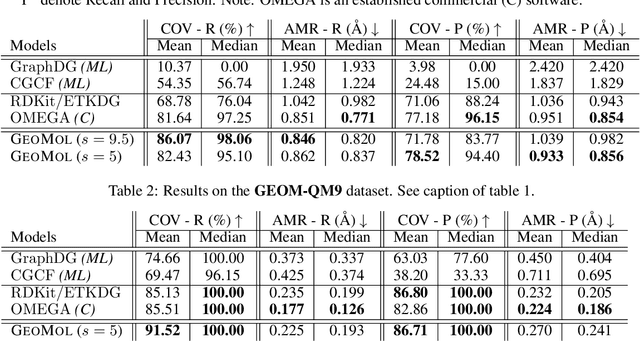
Prediction of a molecule's 3D conformer ensemble from the molecular graph holds a key role in areas of cheminformatics and drug discovery. Existing generative models have several drawbacks including lack of modeling important molecular geometry elements (e.g. torsion angles), separate optimization stages prone to error accumulation, and the need for structure fine-tuning based on approximate classical force-fields or computationally expensive methods such as metadynamics with approximate quantum mechanics calculations at each geometry. We propose GeoMol--an end-to-end, non-autoregressive and SE(3)-invariant machine learning approach to generate distributions of low-energy molecular 3D conformers. Leveraging the power of message passing neural networks (MPNNs) to capture local and global graph information, we predict local atomic 3D structures and torsion angles, avoiding unnecessary over-parameterization of the geometric degrees of freedom (e.g. one angle per non-terminal bond). Such local predictions suffice both for the training loss computation, as well as for the full deterministic conformer assembly (at test time). We devise a non-adversarial optimal transport based loss function to promote diverse conformer generation. GeoMol predominantly outperforms popular open-source, commercial, or state-of-the-art machine learning (ML) models, while achieving significant speed-ups. We expect such differentiable 3D structure generators to significantly impact molecular modeling and related applications.
Message Passing Networks for Molecules with Tetrahedral Chirality
Dec 04, 2020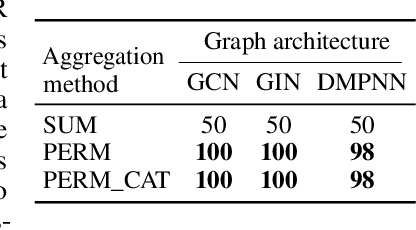
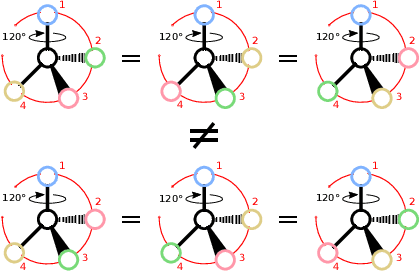
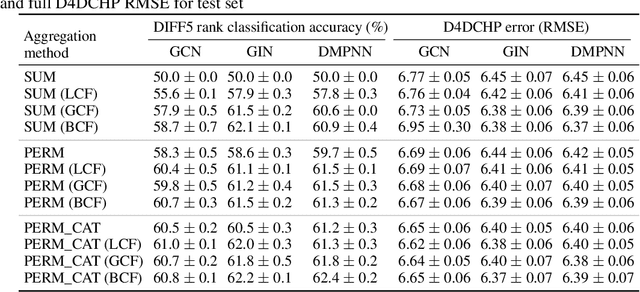
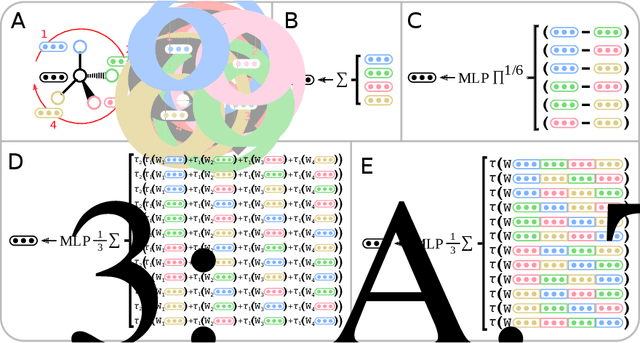
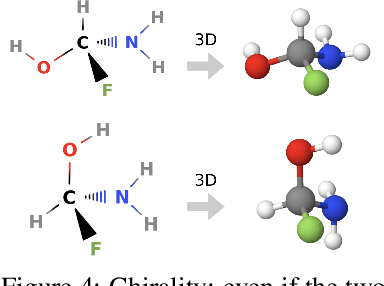
Molecules with identical graph connectivity can exhibit different physical and biological properties if they exhibit stereochemistry-a spatial structural characteristic. However, modern neural architectures designed for learning structure-property relationships from molecular structures treat molecules as graph-structured data and therefore are invariant to stereochemistry. Here, we develop two custom aggregation functions for message passing neural networks to learn properties of molecules with tetrahedral chirality, one common form of stereochemistry. We evaluate performance on synthetic data as well as a newly-proposed protein-ligand docking dataset with relevance to drug discovery. Results show modest improvements over a baseline sum aggregator, highlighting opportunities for further architecture development.