 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeASKCOS: an open source software suite for synthesis planning
Jan 03, 2025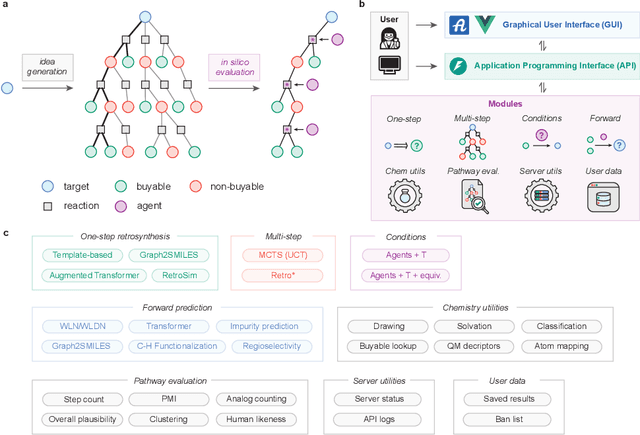



The advancement of machine learning and the availability of large-scale reaction datasets have accelerated the development of data-driven models for computer-aided synthesis planning (CASP) in the past decade. Here, we detail the newest version of ASKCOS, an open source software suite for synthesis planning that makes available several research advances in a freely available, practical tool. Four one-step retrosynthesis models form the basis of both interactive planning and automatic planning modes. Retrosynthetic planning is complemented by other modules for feasibility assessment and pathway evaluation, including reaction condition recommendation, reaction outcome prediction, and auxiliary capabilities such as solubility prediction and quantum mechanical descriptor prediction. ASKCOS has assisted hundreds of medicinal, synthetic, and process chemists in their day-to-day tasks, complementing expert decision making. It is our belief that CASP tools like ASKCOS are an important part of modern chemistry research, and that they offer ever-increasing utility and accessibility.
GeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles
Jun 08, 2021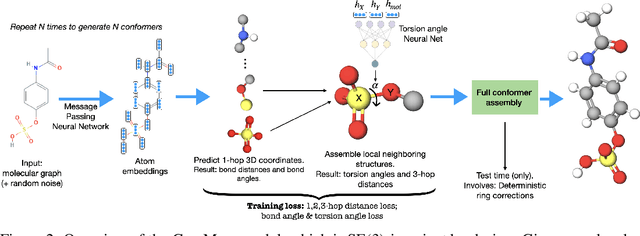
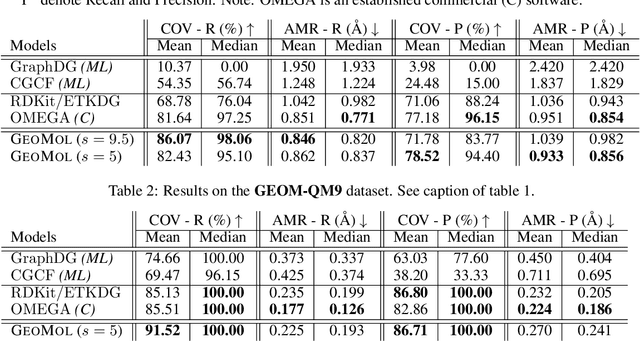
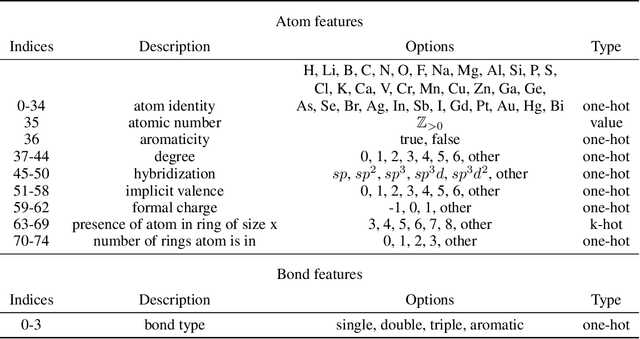
Prediction of a molecule's 3D conformer ensemble from the molecular graph holds a key role in areas of cheminformatics and drug discovery. Existing generative models have several drawbacks including lack of modeling important molecular geometry elements (e.g. torsion angles), separate optimization stages prone to error accumulation, and the need for structure fine-tuning based on approximate classical force-fields or computationally expensive methods such as metadynamics with approximate quantum mechanics calculations at each geometry. We propose GeoMol--an end-to-end, non-autoregressive and SE(3)-invariant machine learning approach to generate distributions of low-energy molecular 3D conformers. Leveraging the power of message passing neural networks (MPNNs) to capture local and global graph information, we predict local atomic 3D structures and torsion angles, avoiding unnecessary over-parameterization of the geometric degrees of freedom (e.g. one angle per non-terminal bond). Such local predictions suffice both for the training loss computation, as well as for the full deterministic conformer assembly (at test time). We devise a non-adversarial optimal transport based loss function to promote diverse conformer generation. GeoMol predominantly outperforms popular open-source, commercial, or state-of-the-art machine learning (ML) models, while achieving significant speed-ups. We expect such differentiable 3D structure generators to significantly impact molecular modeling and related applications.
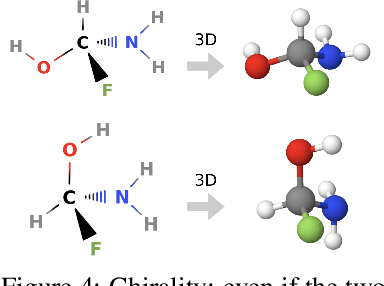
Message Passing Networks for Molecules with Tetrahedral Chirality
Dec 04, 2020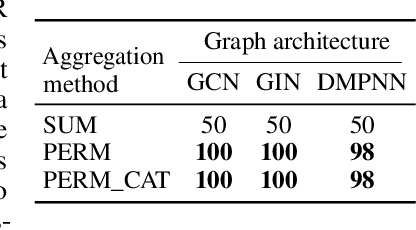
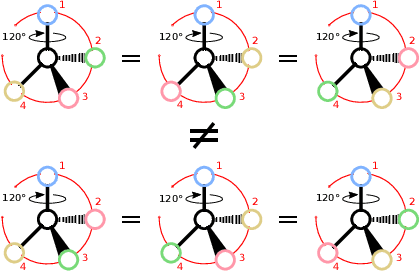
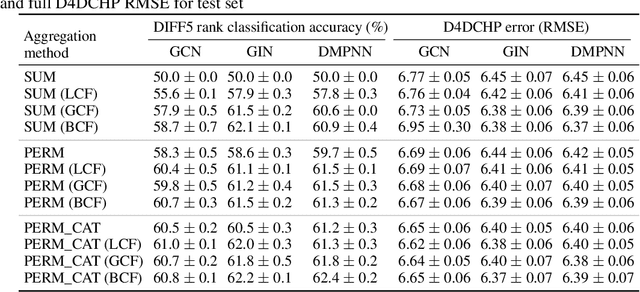
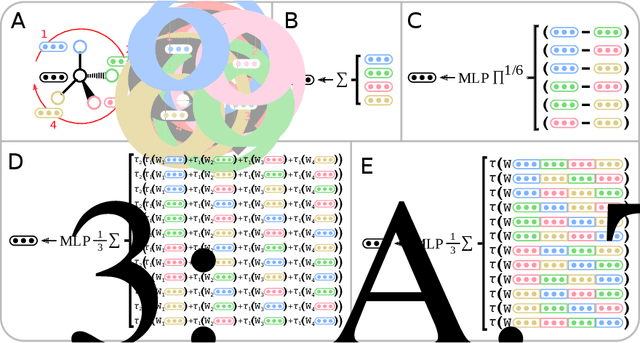
Molecules with identical graph connectivity can exhibit different physical and biological properties if they exhibit stereochemistry-a spatial structural characteristic. However, modern neural architectures designed for learning structure-property relationships from molecular structures treat molecules as graph-structured data and therefore are invariant to stereochemistry. Here, we develop two custom aggregation functions for message passing neural networks to learn properties of molecules with tetrahedral chirality, one common form of stereochemistry. We evaluate performance on synthetic data as well as a newly-proposed protein-ligand docking dataset with relevance to drug discovery. Results show modest improvements over a baseline sum aggregator, highlighting opportunities for further architecture development.
Evaluating Scalable Uncertainty Estimation Methods for DNN-Based Molecular Property Prediction
Oct 07, 2019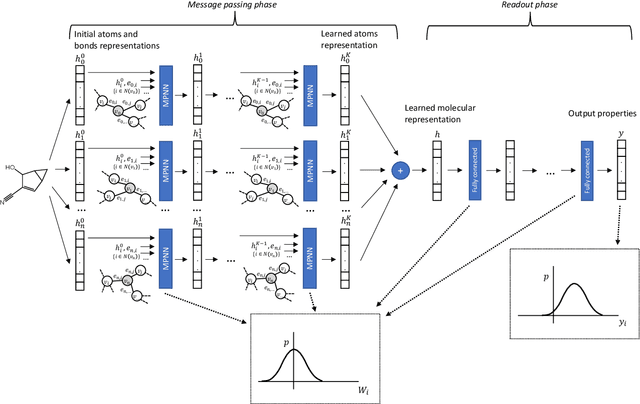

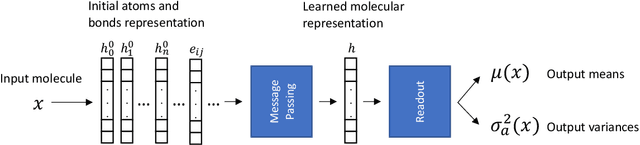
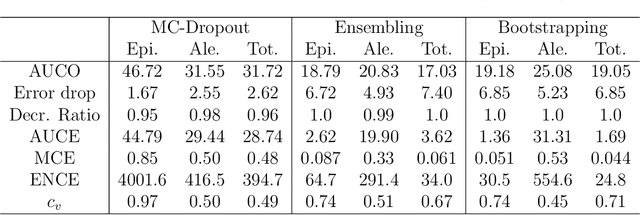
Advances in deep neural network (DNN) based molecular property prediction have recently led to the development of models of remarkable accuracy and generalization ability, with graph convolution neural networks (GCNNs) reporting state-of-the-art performance for this task. However, some challenges remain and one of the most important that needs to be fully addressed concerns uncertainty quantification. DNN performance is affected by the volume and the quality of the training samples. Therefore, establishing when and to what extent a prediction can be considered reliable is just as important as outputting accurate predictions, especially when out-of-domain molecules are targeted. Recently, several methods to account for uncertainty in DNNs have been proposed, most of which are based on approximate Bayesian inference. Among these, only a few scale to the large datasets required in applications. Evaluating and comparing these methods has recently attracted great interest, but results are generally fragmented and absent for molecular property prediction. In this paper, we aim to quantitatively compare scalable techniques for uncertainty estimation in GCNNs. We introduce a set of quantitative criteria to capture different uncertainty aspects, and then use these criteria to compare MC-Dropout, deep ensembles, and bootstrapping, both theoretically in a unified framework that separates aleatoric/epistemic uncertainty and experimentally on the QM9 dataset. Our experiments quantify the performance of the different uncertainty estimation methods and their impact on uncertainty-related error reduction. Our findings indicate that ensembling and bootstrapping consistently outperform MC-Dropout, with different context-specific pros and cons. Our analysis also leads to a better understanding of the role of aleatoric/epistemic uncertainty and highlights the challenge posed by out-of-domain uncertainty.