 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles
Paper and Code
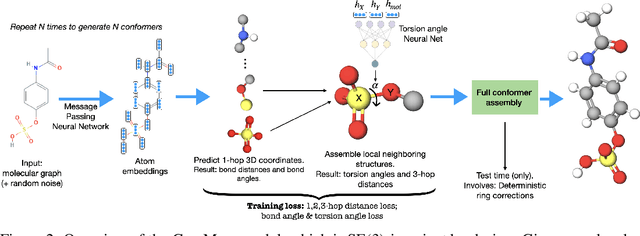
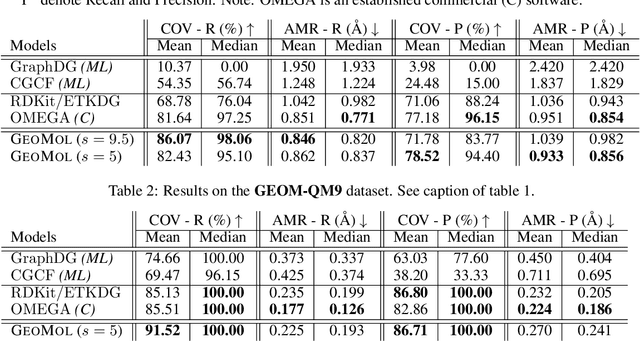
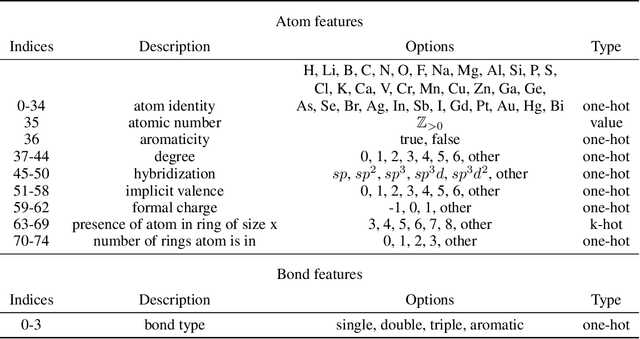
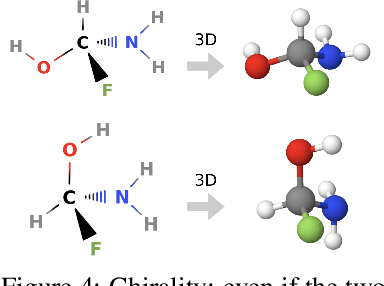
Prediction of a molecule's 3D conformer ensemble from the molecular graph holds a key role in areas of cheminformatics and drug discovery. Existing generative models have several drawbacks including lack of modeling important molecular geometry elements (e.g. torsion angles), separate optimization stages prone to error accumulation, and the need for structure fine-tuning based on approximate classical force-fields or computationally expensive methods such as metadynamics with approximate quantum mechanics calculations at each geometry. We propose GeoMol--an end-to-end, non-autoregressive and SE(3)-invariant machine learning approach to generate distributions of low-energy molecular 3D conformers. Leveraging the power of message passing neural networks (MPNNs) to capture local and global graph information, we predict local atomic 3D structures and torsion angles, avoiding unnecessary over-parameterization of the geometric degrees of freedom (e.g. one angle per non-terminal bond). Such local predictions suffice both for the training loss computation, as well as for the full deterministic conformer assembly (at test time). We devise a non-adversarial optimal transport based loss function to promote diverse conformer generation. GeoMol predominantly outperforms popular open-source, commercial, or state-of-the-art machine learning (ML) models, while achieving significant speed-ups. We expect such differentiable 3D structure generators to significantly impact molecular modeling and related applications.