 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeASKCOS: an open source software suite for synthesis planning
Jan 03, 2025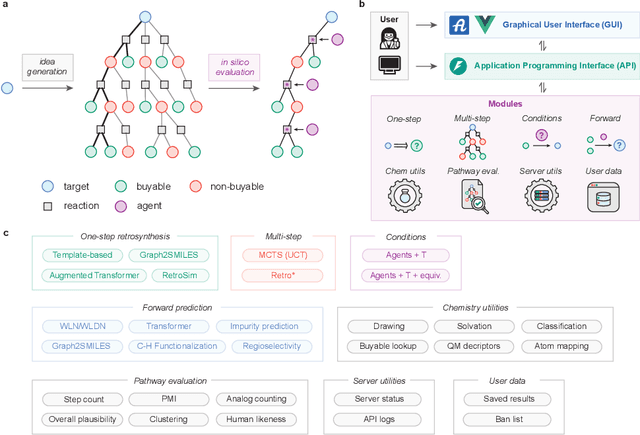



The advancement of machine learning and the availability of large-scale reaction datasets have accelerated the development of data-driven models for computer-aided synthesis planning (CASP) in the past decade. Here, we detail the newest version of ASKCOS, an open source software suite for synthesis planning that makes available several research advances in a freely available, practical tool. Four one-step retrosynthesis models form the basis of both interactive planning and automatic planning modes. Retrosynthetic planning is complemented by other modules for feasibility assessment and pathway evaluation, including reaction condition recommendation, reaction outcome prediction, and auxiliary capabilities such as solubility prediction and quantum mechanical descriptor prediction. ASKCOS has assisted hundreds of medicinal, synthetic, and process chemists in their day-to-day tasks, complementing expert decision making. It is our belief that CASP tools like ASKCOS are an important part of modern chemistry research, and that they offer ever-increasing utility and accessibility.
GeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles
Jun 08, 2021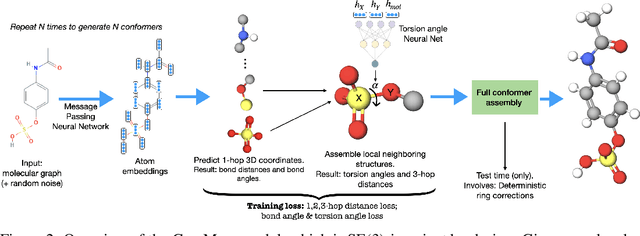
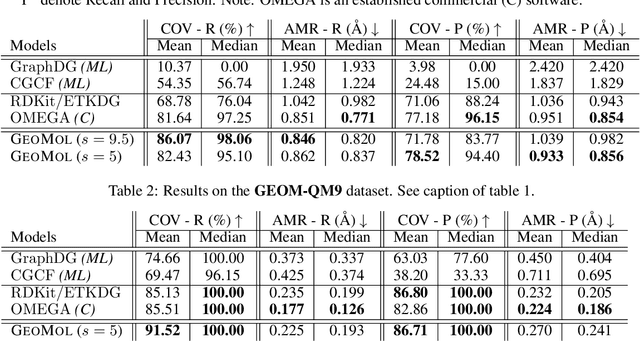
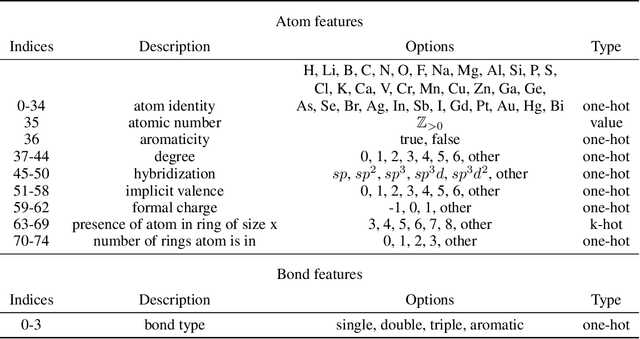
Prediction of a molecule's 3D conformer ensemble from the molecular graph holds a key role in areas of cheminformatics and drug discovery. Existing generative models have several drawbacks including lack of modeling important molecular geometry elements (e.g. torsion angles), separate optimization stages prone to error accumulation, and the need for structure fine-tuning based on approximate classical force-fields or computationally expensive methods such as metadynamics with approximate quantum mechanics calculations at each geometry. We propose GeoMol--an end-to-end, non-autoregressive and SE(3)-invariant machine learning approach to generate distributions of low-energy molecular 3D conformers. Leveraging the power of message passing neural networks (MPNNs) to capture local and global graph information, we predict local atomic 3D structures and torsion angles, avoiding unnecessary over-parameterization of the geometric degrees of freedom (e.g. one angle per non-terminal bond). Such local predictions suffice both for the training loss computation, as well as for the full deterministic conformer assembly (at test time). We devise a non-adversarial optimal transport based loss function to promote diverse conformer generation. GeoMol predominantly outperforms popular open-source, commercial, or state-of-the-art machine learning (ML) models, while achieving significant speed-ups. We expect such differentiable 3D structure generators to significantly impact molecular modeling and related applications.
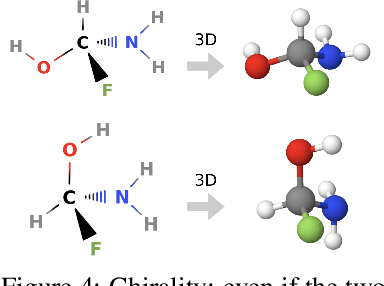
Message Passing Networks for Molecules with Tetrahedral Chirality
Dec 04, 2020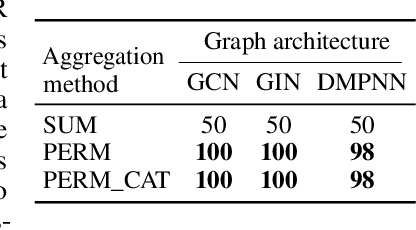
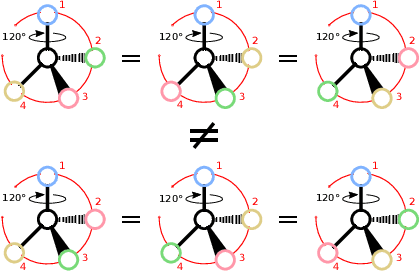
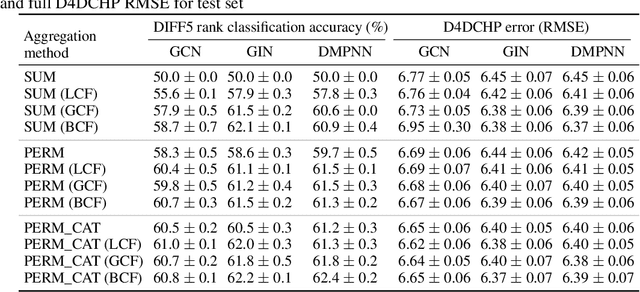
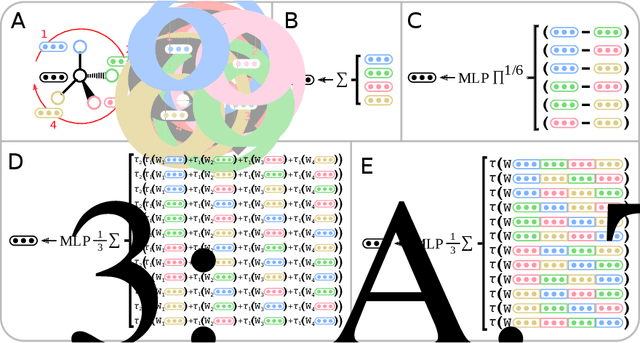
Molecules with identical graph connectivity can exhibit different physical and biological properties if they exhibit stereochemistry-a spatial structural characteristic. However, modern neural architectures designed for learning structure-property relationships from molecular structures treat molecules as graph-structured data and therefore are invariant to stereochemistry. Here, we develop two custom aggregation functions for message passing neural networks to learn properties of molecules with tetrahedral chirality, one common form of stereochemistry. We evaluate performance on synthetic data as well as a newly-proposed protein-ligand docking dataset with relevance to drug discovery. Results show modest improvements over a baseline sum aggregator, highlighting opportunities for further architecture development.
Autonomous discovery in the chemical sciences part II: Outlook
Mar 30, 2020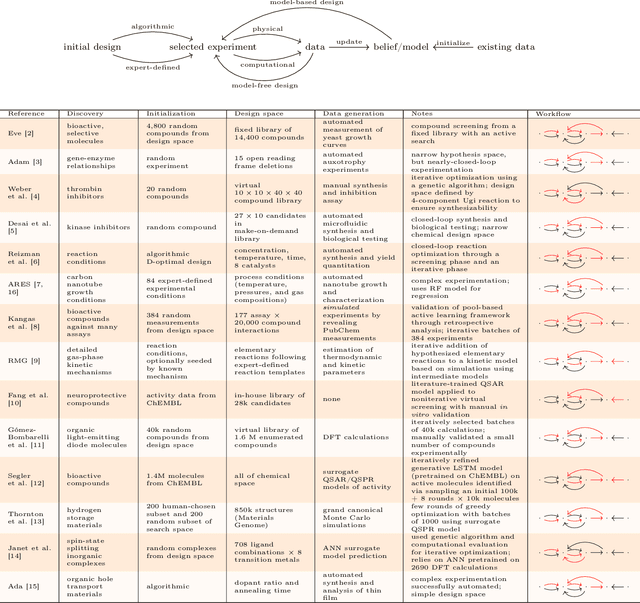
This two-part review examines how automation has contributed to different aspects of discovery in the chemical sciences. In this second part, we reflect on a selection of exemplary studies. It is increasingly important to articulate what the role of automation and computation has been in the scientific process and how that has or has not accelerated discovery. One can argue that even the best automated systems have yet to ``discover'' despite being incredibly useful as laboratory assistants. We must carefully consider how they have been and can be applied to future problems of chemical discovery in order to effectively design and interact with future autonomous platforms. The majority of this article defines a large set of open research directions, including improving our ability to work with complex data, build empirical models, automate both physical and computational experiments for validation, select experiments, and evaluate whether we are making progress toward the ultimate goal of autonomous discovery. Addressing these practical and methodological challenges will greatly advance the extent to which autonomous systems can make meaningful discoveries.
Autonomous discovery in the chemical sciences part I: Progress
Mar 30, 2020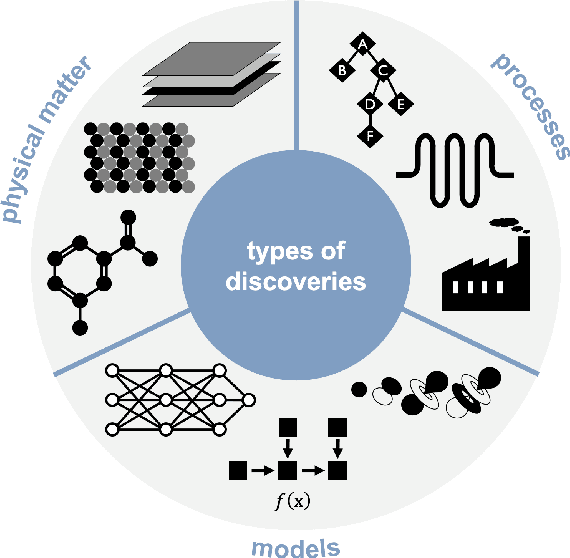


This two-part review examines how automation has contributed to different aspects of discovery in the chemical sciences. In this first part, we describe a classification for discoveries of physical matter (molecules, materials, devices), processes, and models and how they are unified as search problems. We then introduce a set of questions and considerations relevant to assessing the extent of autonomy. Finally, we describe many case studies of discoveries accelerated by or resulting from computer assistance and automation from the domains of synthetic chemistry, drug discovery, inorganic chemistry, and materials science. These illustrate how rapid advancements in hardware automation and machine learning continue to transform the nature of experimentation and modelling. Part two reflects on these case studies and identifies a set of open challenges for the field.