 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSimilarity-Quantized Relative Difference Learning for Improved Molecular Activity Prediction
Jan 15, 2025



Accurate prediction of molecular activities is crucial for efficient drug discovery, yet remains challenging due to limited and noisy datasets. We introduce Similarity-Quantized Relative Learning (SQRL), a learning framework that reformulates molecular activity prediction as relative difference learning between structurally similar pairs of compounds. SQRL uses precomputed molecular similarities to enhance training of graph neural networks and other architectures, and significantly improves accuracy and generalization in low-data regimes common in drug discovery. We demonstrate its broad applicability and real-world potential through benchmarking on public datasets as well as proprietary industry data. Our findings demonstrate that leveraging similarity-aware relative differences provides an effective paradigm for molecular activity prediction.
RINGER: Rapid Conformer Generation for Macrocycles with Sequence-Conditioned Internal Coordinate Diffusion
May 30, 2023



Macrocyclic peptides are an emerging therapeutic modality, yet computational approaches for accurately sampling their diverse 3D ensembles remain challenging due to their conformational diversity and geometric constraints. Here, we introduce RINGER, a diffusion-based transformer model for sequence-conditioned generation of macrocycle structures based on internal coordinates. RINGER provides fast backbone sampling while respecting key structural invariances of cyclic peptides. Through extensive benchmarking and analysis against gold-standard conformer ensembles of cyclic peptides generated with metadynamics, we demonstrate how RINGER generates both high-quality and diverse geometries at a fraction of the computational cost. Our work lays the foundation for improved sampling of cyclic geometries and the development of geometric learning methods for peptides.
CREMP: Conformer-Rotamer Ensembles of Macrocyclic Peptides for Machine Learning
May 14, 2023Computational and machine learning approaches to model the conformational landscape of macrocyclic peptides have the potential to enable rational design and optimization. However, accurate, fast, and scalable methods for modeling macrocycle geometries remain elusive. Recent deep learning approaches have significantly accelerated protein structure prediction and the generation of small-molecule conformational ensembles, yet similar progress has not been made for macrocyclic peptides due to their unique properties. Here, we introduce CREMP, a resource generated for the rapid development and evaluation of machine learning models for macrocyclic peptides. CREMP contains 36,198 unique macrocyclic peptides and their high-quality structural ensembles generated using the Conformer-Rotamer Ensemble Sampling Tool (CREST). Altogether, this new dataset contains nearly 31.3 million unique macrocycle geometries, each annotated with energies derived from semi-empirical extended tight-binding (xTB) DFT calculations. We anticipate that this dataset will enable the development of machine learning models that can improve peptide design and optimization for novel therapeutics.
Evaluating Scalable Uncertainty Estimation Methods for DNN-Based Molecular Property Prediction
Oct 07, 2019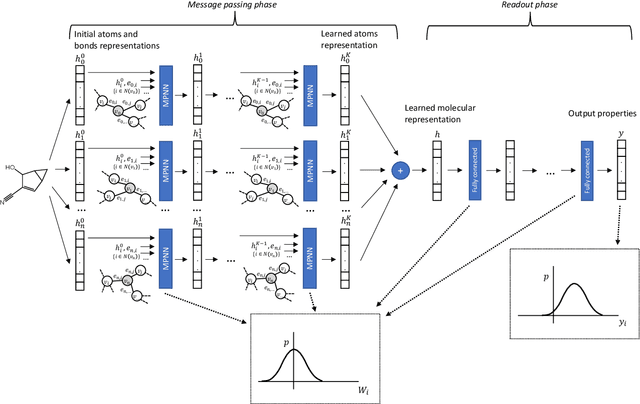

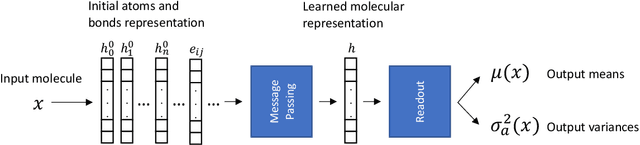
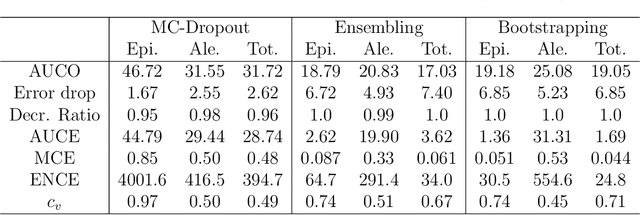
Advances in deep neural network (DNN) based molecular property prediction have recently led to the development of models of remarkable accuracy and generalization ability, with graph convolution neural networks (GCNNs) reporting state-of-the-art performance for this task. However, some challenges remain and one of the most important that needs to be fully addressed concerns uncertainty quantification. DNN performance is affected by the volume and the quality of the training samples. Therefore, establishing when and to what extent a prediction can be considered reliable is just as important as outputting accurate predictions, especially when out-of-domain molecules are targeted. Recently, several methods to account for uncertainty in DNNs have been proposed, most of which are based on approximate Bayesian inference. Among these, only a few scale to the large datasets required in applications. Evaluating and comparing these methods has recently attracted great interest, but results are generally fragmented and absent for molecular property prediction. In this paper, we aim to quantitatively compare scalable techniques for uncertainty estimation in GCNNs. We introduce a set of quantitative criteria to capture different uncertainty aspects, and then use these criteria to compare MC-Dropout, deep ensembles, and bootstrapping, both theoretically in a unified framework that separates aleatoric/epistemic uncertainty and experimentally on the QM9 dataset. Our experiments quantify the performance of the different uncertainty estimation methods and their impact on uncertainty-related error reduction. Our findings indicate that ensembling and bootstrapping consistently outperform MC-Dropout, with different context-specific pros and cons. Our analysis also leads to a better understanding of the role of aleatoric/epistemic uncertainty and highlights the challenge posed by out-of-domain uncertainty.