 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning 3D Representations of Molecular Chirality with Invariance to Bond Rotations
Paper and Code
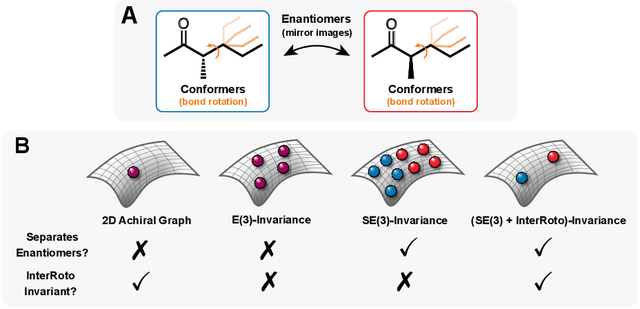

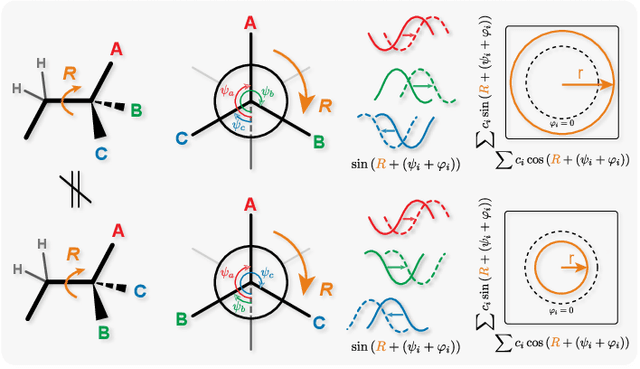
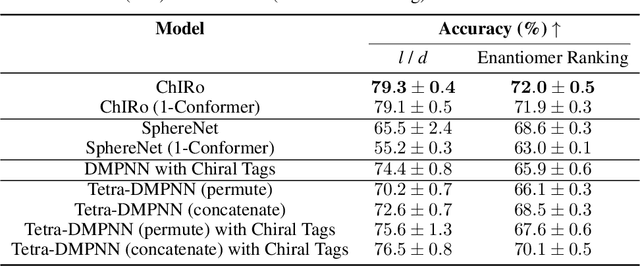
Molecular chirality, a form of stereochemistry most often describing relative spatial arrangements of bonded neighbors around tetrahedral carbon centers, influences the set of 3D conformers accessible to the molecule without changing its 2D graph connectivity. Chirality can strongly alter (bio)chemical interactions, particularly protein-drug binding. Most 2D graph neural networks (GNNs) designed for molecular property prediction at best use atomic labels to na\"ively treat chirality, while E(3)-invariant 3D GNNs are invariant to chirality altogether. To enable representation learning on molecules with defined stereochemistry, we design an SE(3)-invariant model that processes torsion angles of a 3D molecular conformer. We explicitly model conformational flexibility by integrating a novel type of invariance to rotations about internal molecular bonds into the architecture, mitigating the need for multi-conformer data augmentation. We test our model on four benchmarks: contrastive learning to distinguish conformers of different stereoisomers in a learned latent space, classification of chiral centers as R/S, prediction of how enantiomers rotate circularly polarized light, and ranking enantiomers by their docking scores in an enantiosensitive protein pocket. We compare our model, Chiral InterRoto-Invariant Neural Network (ChIRo), with 2D and 3D GNNs to demonstrate that our model achieves state of the art performance when learning chiral-sensitive functions from molecular structures.