 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReactive Flux Matching: Mechanism Discovery and Adaptive Sampling of Rare Events
Jun 04, 2026Path sampling methods generate ensembles of reactive trajectories connecting metastable states, but extracting mechanistic insight from these data remains nontrivial. We introduce Flux Matching, a framework that learns two complementary objects directly from reactive trajectory data: a current velocity $u(z)$, whose streamlines trace the dominant reaction pathways, and a scalar potential $h(z)$, obtained from a weighted Helmholtz-Hodge decomposition of the reactive current, that serves as a data-driven reaction coordinate. Both minimize quadratic functionals over the reactive path ensemble, analogous to the flow matching loss in generative modeling, and require no knowledge of the underlying dynamics or stationary distribution. Unlike committor-based methods, $u$ and $h$ remain well-defined under projection onto non-Markovian collective variables, and their level sets in turn provide adaptive interfaces for improved sampling with enhanced sampling methods. Flux Matching is validated through the generation of current velocity trajectories and rate constant calculations on molecular systems.
Scalable Inference-Time Annealing with Surrogate Likelihood Estimators
Jun 01, 2026A long standing challenge in computational chemistry and biophysics is efficiently sampling the Boltzmann distribution of molecules. Advances in generative modeling have been proposed to address the limitations of conventional sampling techniques by eliminating the computational cost of simulation. A promising direction is iteratively finetuning diffusion models along a temperature ladder whereby training data is generated via importance sampling during inference-time annealing. Unfortunately, these methods require computing a divergence over the score field to estimate importance weights, rendering them intractable for larger systems. Here we present scalable inference-time annealing (SITA), which retrains flow-based models to generate samples at progressively lower temperatures using an energy-based model to facilitate fast surrogate likelihoods. We demonstrate state-of-the-art performance on both Alanine Dipeptide and Alanine Tripeptide while avoiding costly divergence terms. Our code is available at https://github.com/countrsignal/sita.git
BoltzNCE: Learning Likelihoods for Boltzmann Generation with Stochastic Interpolants and Noise Contrastive Estimation
Jul 02, 2025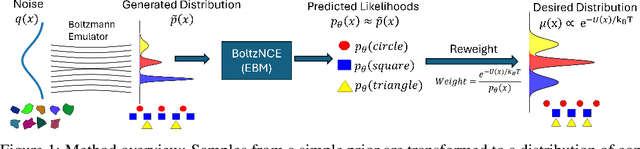

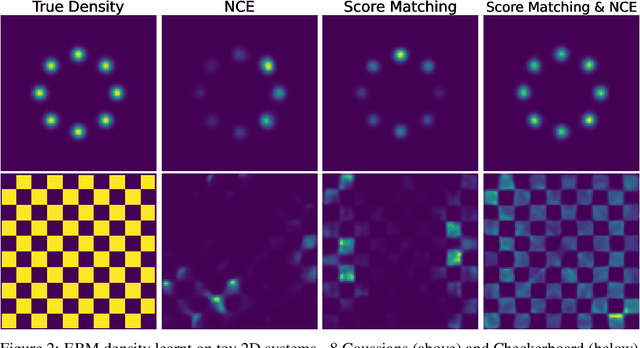
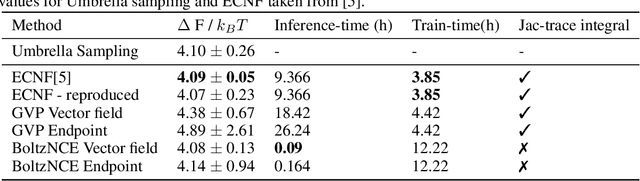
Efficient sampling from the Boltzmann distribution defined by an energy function is a key challenge in modeling physical systems such as molecules. Boltzmann Generators tackle this by leveraging Continuous Normalizing Flows that transform a simple prior into a distribution that can be reweighted to match the Boltzmann distribution using sample likelihoods. However, obtaining likelihoods requires computing costly Jacobians during integration, making it impractical for large molecular systems. To overcome this, we propose learning the likelihood of the generated distribution via an energy-based model trained with noise contrastive estimation and score matching. By using stochastic interpolants to anneal between the prior and generated distributions, we combine both the objective functions to efficiently learn the density function. On the alanine dipeptide system, we demonstrate that our method yields free energy profiles and energy distributions comparable to those obtained with exact likelihoods. Additionally, we show that free energy differences between metastable states can be estimated accurately with orders-of-magnitude speedup.
GEOM-Drugs Revisited: Toward More Chemically Accurate Benchmarks for 3D Molecule Generation
Apr 30, 2025



Deep generative models have shown significant promise in generating valid 3D molecular structures, with the GEOM-Drugs dataset serving as a key benchmark. However, current evaluation protocols suffer from critical flaws, including incorrect valency definitions, bugs in bond order calculations, and reliance on force fields inconsistent with the reference data. In this work, we revisit GEOM-Drugs and propose a corrected evaluation framework: we identify and fix issues in data preprocessing, construct chemically accurate valency tables, and introduce a GFN2-xTB-based geometry and energy benchmark. We retrain and re-evaluate several leading models under this framework, providing updated performance metrics and practical recommendations for future benchmarking. Our results underscore the need for chemically rigorous evaluation practices in 3D molecular generation. Our recommended evaluation methods and GEOM-Drugs processing scripts are available at https://github.com/isayevlab/geom-drugs-3dgen-evaluation.
Mixed Continuous and Categorical Flow Matching for 3D De Novo Molecule Generation
Apr 30, 2024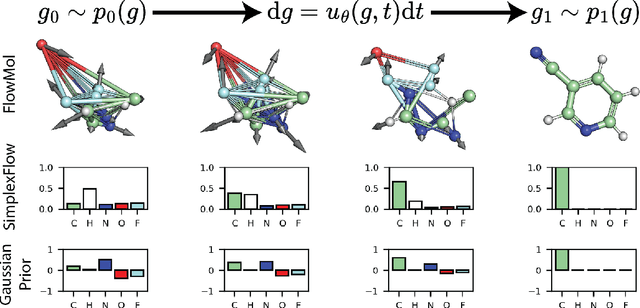
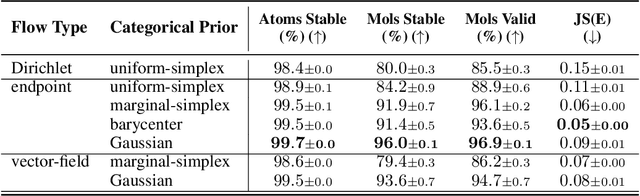
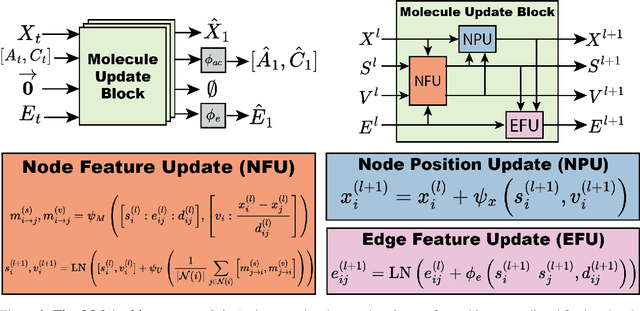
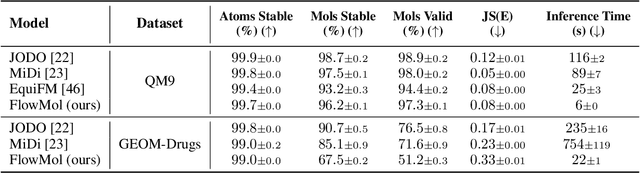
Deep generative models that produce novel molecular structures have the potential to facilitate chemical discovery. Diffusion models currently achieve state of the art performance for 3D molecule generation. In this work, we explore the use of flow matching, a recently proposed generative modeling framework that generalizes diffusion models, for the task of de novo molecule generation. Flow matching provides flexibility in model design; however, the framework is predicated on the assumption of continuously-valued data. 3D de novo molecule generation requires jointly sampling continuous and categorical variables such as atom position and atom type. We extend the flow matching framework to categorical data by constructing flows that are constrained to exist on a continuous representation of categorical data known as the probability simplex. We call this extension SimplexFlow. We explore the use of SimplexFlow for de novo molecule generation. However, we find that, in practice, a simpler approach that makes no accommodations for the categorical nature of the data yields equivalent or superior performance. As a result of these experiments, we present FlowMol, a flow matching model for 3D de novo generative model that achieves improved performance over prior flow matching methods, and we raise important questions about the design of prior distributions for achieving strong performance in flow matching models. Code and trained models for reproducing this work are available at https://github.com/dunni3/FlowMol
Accelerating Inference in Molecular Diffusion Models with Latent Representations of Protein Structure
Nov 22, 2023



Diffusion generative models have emerged as a powerful framework for addressing problems in structural biology and structure-based drug design. These models operate directly on 3D molecular structures. Due to the unfavorable scaling of graph neural networks (GNNs) with graph size as well as the relatively slow inference speeds inherent to diffusion models, many existing molecular diffusion models rely on coarse-grained representations of protein structure to make training and inference feasible. However, such coarse-grained representations discard essential information for modeling molecular interactions and impair the quality of generated structures. In this work, we present a novel GNN-based architecture for learning latent representations of molecular structure. When trained end-to-end with a diffusion model for de novo ligand design, our model achieves comparable performance to one with an all-atom protein representation while exhibiting a 3-fold reduction in inference time.
Generating 3D Molecules Conditional on Receptor Binding Sites with Deep Generative Models
Oct 28, 2021
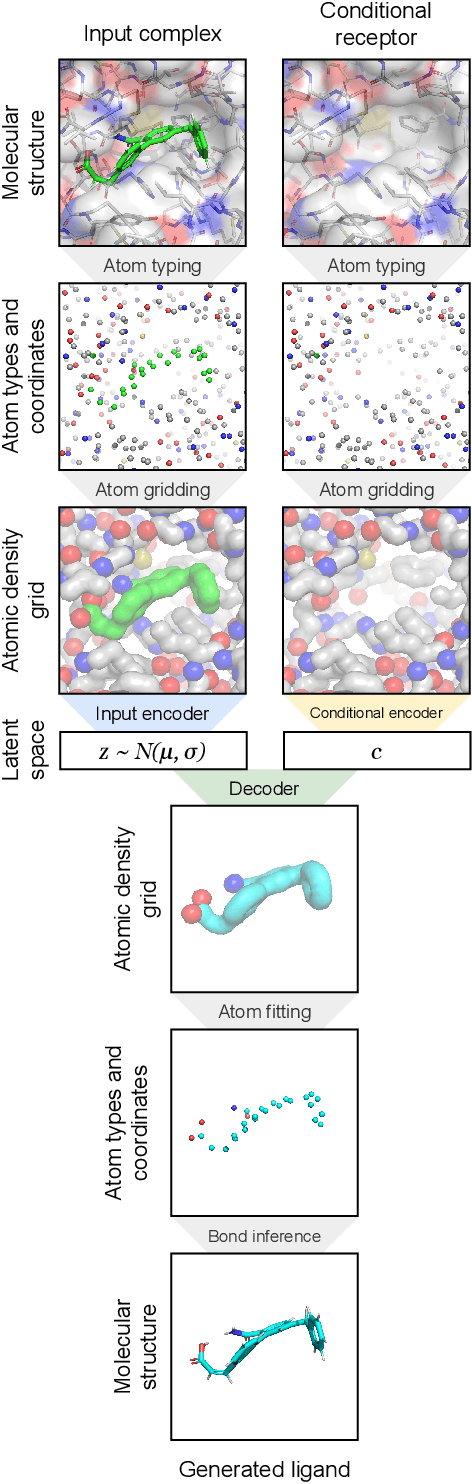
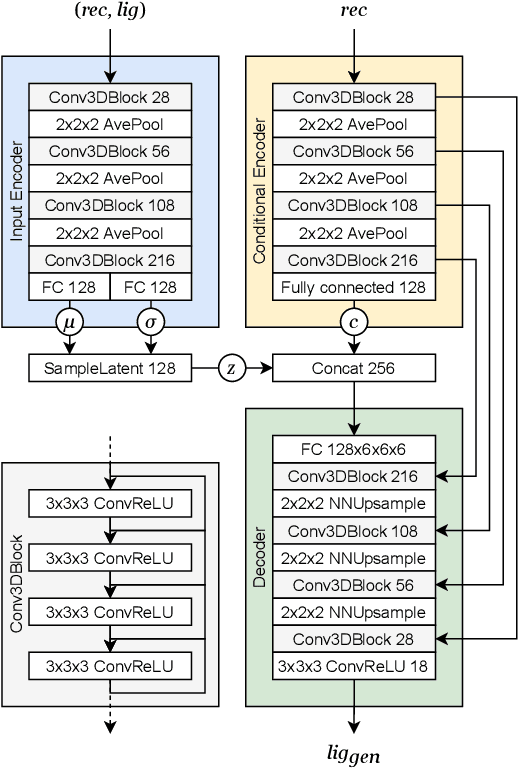
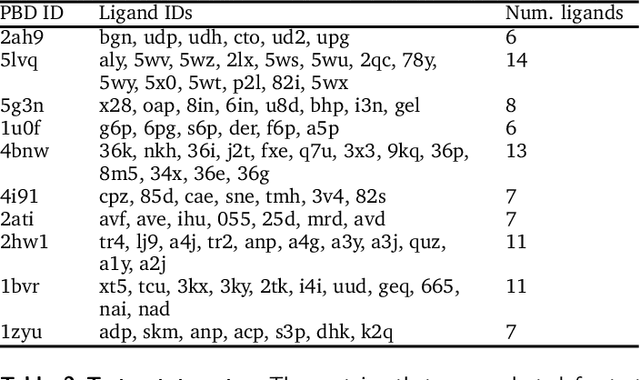
The goal of structure-based drug discovery is to find small molecules that bind to a given target protein. Deep learning has been used to generate drug-like molecules with certain cheminformatic properties, but has not yet been applied to generating 3D molecules predicted to bind to proteins by sampling the conditional distribution of protein-ligand binding interactions. In this work, we describe for the first time a deep learning system for generating 3D molecular structures conditioned on a receptor binding site. We approach the problem using a conditional variational autoencoder trained on an atomic density grid representation of cross-docked protein-ligand structures. We apply atom fitting and bond inference procedures to construct valid molecular conformations from generated atomic densities. We evaluate the properties of the generated molecules and demonstrate that they change significantly when conditioned on mutated receptors. We also explore the latent space learned by our generative model using sampling and interpolation techniques. This work opens the door for end-to-end prediction of stable bioactive molecules from protein structures with deep learning.
Learning a Continuous Representation of 3D Molecular Structures with Deep Generative Models
Oct 20, 2020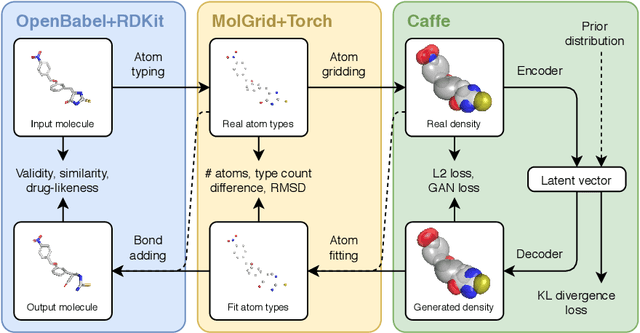
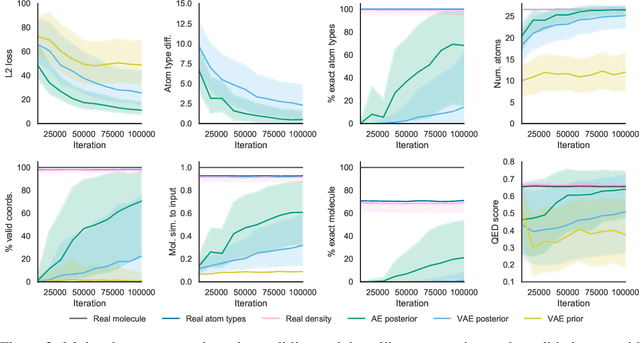
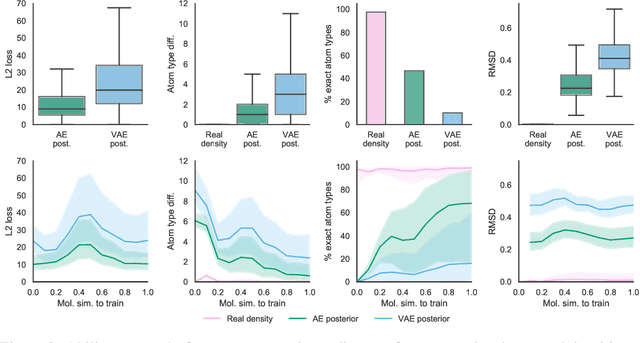
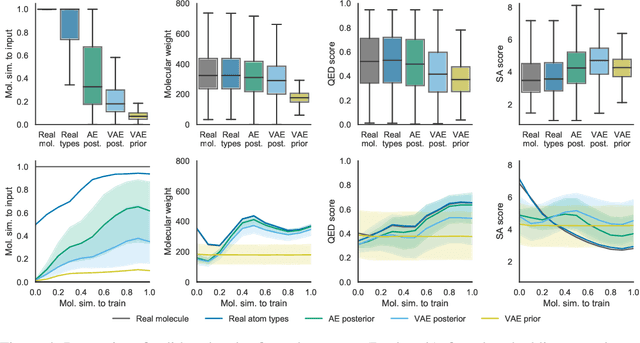
Machine learning methods in drug discovery have primarily focused on virtual screening of molecular libraries using discriminative models. Generative models are an entirely different approach to drug discovery that learn to represent and optimize molecules in a continuous latent space. These methods have already been applied with increasing success to the generation of two dimensional molecules as SMILES strings and molecular graphs. In this work, we describe deep generative models for three dimensional molecular structures using atomic density grids and a novel fitting algorithm that converts continuous grids to discrete molecular structures. Our models jointly represent drug-like molecules and their conformations in a latent space that can be explored through interpolation. We are able to sample diverse sets of molecules based on a given input compound and increase the probability of creating a valid, drug-like molecule.
Generating 3D Molecular Structures Conditional on a Receptor Binding Site with Deep Generative Models
Oct 16, 2020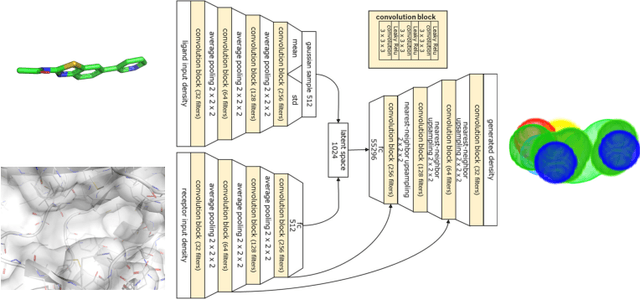
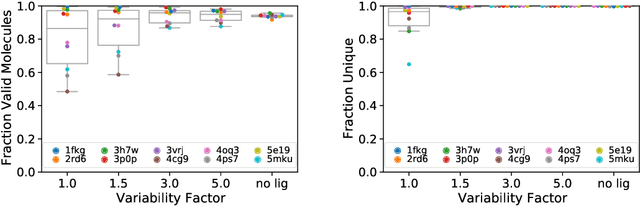
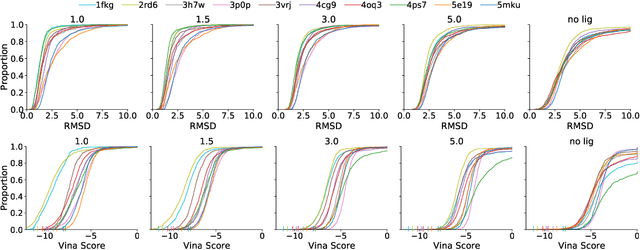
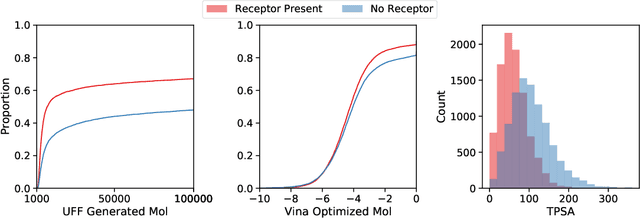
Deep generative models have been applied with increasing success to the generation of two dimensional molecules as SMILES strings and molecular graphs. In this work we describe for the first time a deep generative model that can generate 3D molecular structures conditioned on a three-dimensional (3D) binding pocket. Using convolutional neural networks, we encode atomic density grids into separate receptor and ligand latent spaces. The ligand latent space is variational to support sampling of new molecules. A decoder network generates atomic densities of novel ligands conditioned on the receptor. Discrete atoms are then fit to these continuous densities to create molecular structures. We show that valid and unique molecules can be readily sampled from the variational latent space defined by a reference `seed' structure and generated structures have reasonable interactions with the binding site. As structures are sampled farther in latent space from the seed structure, the novelty of the generated structures increases, but the predicted binding affinity decreases. Overall, we demonstrate the feasibility of conditional 3D molecular structure generation and provide a starting point for methods that also explicitly optimize for desired molecular properties, such as high binding affinity.
SidechainNet: An All-Atom Protein Structure Dataset for Machine Learning
Oct 16, 2020


Despite recent advancements in deep learning methods for protein structure prediction and representation, little focus has been directed at the simultaneous inclusion and prediction of protein backbone and sidechain structure information. We present SidechainNet, a new dataset that directly extends the ProteinNet dataset. SidechainNet includes angle and atomic coordinate information capable of describing all heavy atoms of each protein structure. In this paper, we first provide background information on the availability of protein structure data and the significance of ProteinNet. Thereafter, we argue for the potentially beneficial inclusion of sidechain information through SidechainNet, describe the process by which we organize SidechainNet, and provide a software package (https://github.com/jonathanking/sidechainnet) for data manipulation and training with machine learning models.