 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgelibmolgrid: GPU Accelerated Molecular Gridding for Deep Learning Applications
Dec 10, 2019
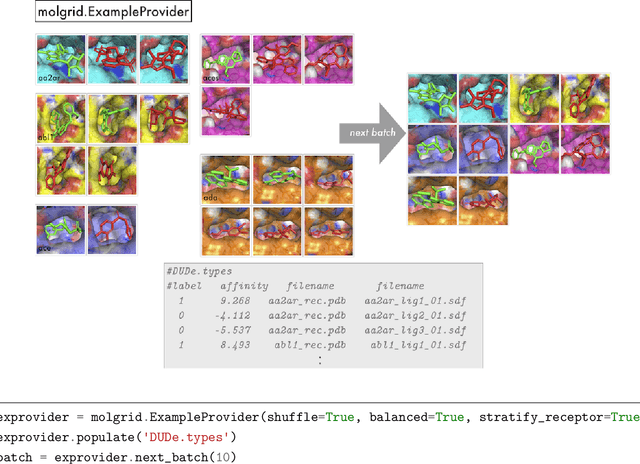

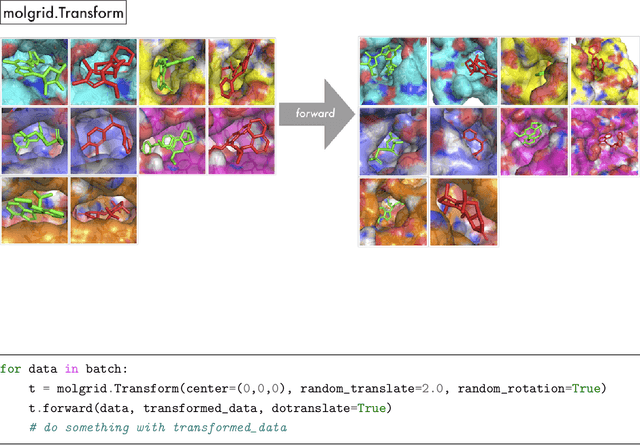
There are many ways to represent a molecule as input to a machine learning model and each is associated with loss and retention of certain kinds of information. In the interest of preserving three-dimensional spatial information, including bond angles and torsions, we have developed libmolgrid, a general-purpose library for representing three-dimensional molecules using multidimensional arrays. This library also provides functionality for composing batches of data suited to machine learning workflows, including data augmentation, class balancing, and example stratification according to a regression variable or data subgroup, and it further supports temporal and spatial recurrences over that data to facilitate work with recurrent neural networks, dynamical data, and size extensive modeling. It was designed for seamless integration with popular deep learning frameworks, including Caffe, PyTorch, and Keras, providing good performance by leveraging graphical processing units (GPUs) for computationally-intensive tasks and efficient memory usage through the use of memory views over preallocated buffers. libmolgrid is a free and open source project that is actively supported, serving the growing need in the molecular modeling community for tools that streamline the process of data ingestion, representation construction, and principled machine learning model development.
Protein-Ligand Scoring with Convolutional Neural Networks
Dec 08, 2016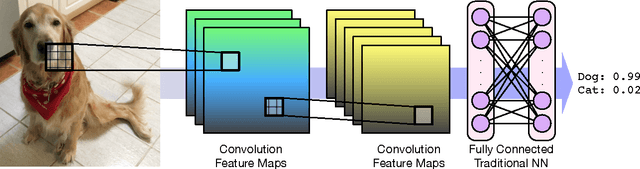

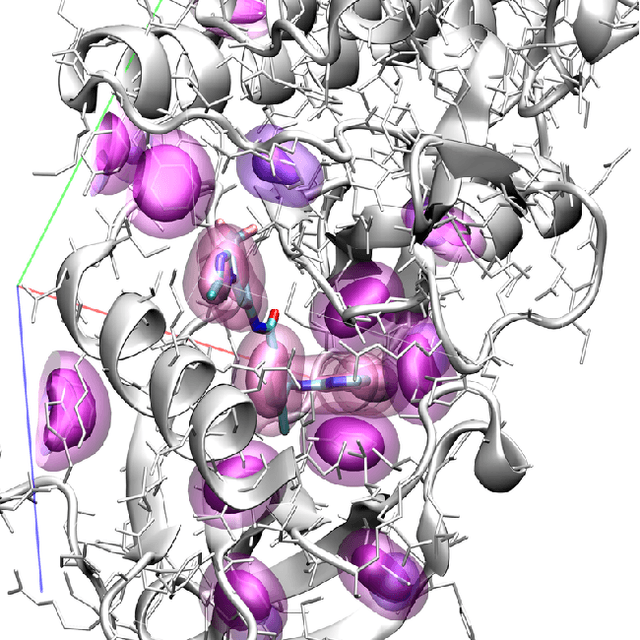
Computational approaches to drug discovery can reduce the time and cost associated with experimental assays and enable the screening of novel chemotypes. Structure-based drug design methods rely on scoring functions to rank and predict binding affinities and poses. The ever-expanding amount of protein-ligand binding and structural data enables the use of deep machine learning techniques for protein-ligand scoring. We describe convolutional neural network (CNN) scoring functions that take as input a comprehensive 3D representation of a protein-ligand interaction. A CNN scoring function automatically learns the key features of protein-ligand interactions that correlate with binding. We train and optimize our CNN scoring functions to discriminate between correct and incorrect binding poses and known binders and non-binders. We find that our CNN scoring function outperforms the AutoDock Vina scoring function when ranking poses both for pose prediction and virtual screening.